Flavin C Vitamin
Introduction to Vitamins and Minerals
Vitamins are organic molecules that function in a wide variety of capacities within the body. The most prominent function of the vitamins is to serve as cofactors (co-enzymes) for enzymatic reactions. The distinguishing feature of the vitamins is that they generally cannot be synthesized by mammalian cells and, therefore, must be supplied in the diet. The vitamins are of two distinct types, water soluble and fat soluble. For more information on the food products that are good sources of the individual vitamins visit the Supplement Science page.
The minerals that are considered of dietary significance are those that are necessary to support biochemical reactions by serving both functional and structural roles as well as those serving as electrolytes. The use of the term dietary mineral is considered archaic since the intent of the term "mineral" is to describe ions not actual minerals. There are both quantity elements required by the body and trace elements. The quantity elements are sodium, magnesium, phosphorous, sulfur, chlorine, potassium and calcium. The essential trace elements are manganese, iron, cobalt, nickel, copper, zinc, selenium, molybdenum, and iodine. Additional trace elements (although not considered essential) are boron, chromium, fluoride, and silicon.
Thiamine (Thiamin)
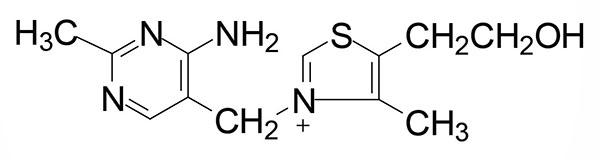
Thiamine (also written thiamin) is also known as vitamin B1. Thiamine is derived from a substituted pyrimidine and a thiazole which are coupled by a methylene bridge. Thiamine is the form of vitamin B1 that is absorbed from the small intestine. Thiamine uptake from the intestines is a function of the solute carrier transporter family member encoded by the SLC19A2 gene. Uptake of thiamine into cells from the blood occurs primarily through the activity of the SLC19A3 encoded transporter. Mitochondrial uptake of activated thiamine (thiamine pyrophosphate: TPP) occurs via the action of the transporter encoded by the SLC25A19 gene.
Mutations in the SLC19A2 gene are associated with thiamine responsive megaloblastic anemia syndrome (TRMA). TRMA is also referred to as thiamine metabolism dysfunction syndrome 1 (THMD1). TRMA is an autosomal recessive disease associated with megaloblastic anemia, diabetes mellitus, and sensorineural deafness. TRMA most often manifests between infancy and adolescence. Improvement in the anemia, and often the diabetes, is seen in patients treated with thiamine, hence the name of the disorder.
Mutations in the SLC19A3 gene are associated with thiamine metabolism dysfunction syndrome 2 (THMD2). THMD2 is also known as basal ganglia disease, biotin responsive (BBGD). THMD2 is an autosomal recessive disease characterized by episodic encephalopathy that is most often triggered by a febrile illness. Additional presenting symptoms include dysphagia (difficulty swallowing), confusion, seizures, and external ophthalmoplegia. The disorder can often lead to coma and death. Administration of high doses of biotin, and sometimes thiamine, has been shown to result in partial or complete improvement within days. The precise role of biotin treatment in THMD2 in unknown since the defect is in the transporter for cellular thiamine uptake. However, it is thought that biotin may lead to increased expression of SLC19A3 gene.
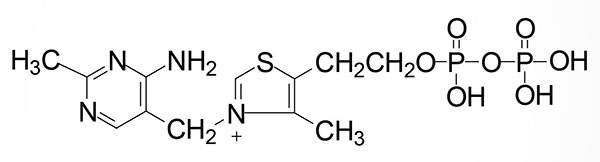
Thiamine is rapidly converted to its active form, thiamine pyrophosphate, TPP, by the enzyme thiamine pyrophosphokinase 1, TPK1. The TPK1 gene is located on chromosome 7q35 and is composed of 27 exons that generate 15 alternatively spliced mRNAs. These TPK1 mRNAs encode a total of six different protein isoforms. Expression of the TPK1 gene is found in all tissues with highest levels in the small intestine.
TPP is necessary as a cofactor for three critical dehydrogenases. These enzymes are the pyruvate dehydrogenase complex (PDHc) and 2-oxoglutarate (α-ketoglutarate) dehydrogenase (OGDH), both of which are associated with the TCA cycle, and branched-chain ketoacid dehydrogenase (BCKD) necessary for metabolism of the branched-chain amino acids, leucine, isoleucine, and valine. These three dehydrogenases also require the co-factors derived from the vitamins lipoic acid, pantothenic acid (CoA), riboflavin, and niacin. For this reason these three dehydrogenases are often referred to as theTender (thiamine)Loving (lipoic acid)Care (CoA)For (flavin)Nancy (niacin) enzymes. In addition to these three dehydrogenases, TPP is a required co-factor for the transketolase catalyzed reactions of the pentose phosphate pathway and it is required for the catabolism of the methyl-substituted fatty acid (phytanic acid) at the 2-hydroxyphytanoyl-CoA lyase reaction. A deficiency in thiamine intake leads to a severely reduced capacity of cells to generate energy and to carry out reductive biosynthetic reactions as well as to synthesize nucleotides because of its role in the three critical dehydrogenase complexes.
Dietary Sources of Thiamine
The dietary requirement for thiamine is proportional to the caloric intake of the diet and ranges from 1.0–1.5 mg/day for normal adults. If the carbohydrate content of the diet is excessive then an increase in thiamine intake will be required.
The richest sources of vitamin B1 include yeasts and animal liver. Additional sources include whole-grain cereals, rye and whole-wheat flour, navy beans, kidney beans, wheat germ, as well as pork and fish.
| Food source | Thiamine content (mg) |
| Yeast, brewer's, 2 tbls | 2.3 |
| Pork chop, lean, 100 gm | 0.9 |
| Ham, lean, 100 gm | 0.7 |
| Catfish, 100 gm cooked | 0.4 |
| Bagel, 2 oz enriched | 0.4 |
| Milk, soy, 130 gm | 0.4 |
| Beans, baked, 130 gm | 0.34 |
| Oatmeal, 130 gm cooked | 0.26 |
| Rice, white, cooked, 130 gm | 0.26 |
| Green peas, 65 gm cooked | 0.23 |
| Potato, one medium baked | 0.22 |
| Orange juice, 130 gm | 0.20 |
| Black beans, 65 gm cooked | 0.21 |
| Navy beans, 65 gm cooked | 0.19 |
| Soy nuts, 65 gm | 0.20 |
| Cashews, 65 gm | 0.15 |
| Peanuts, 65 gm | 0.10 |
Clinical Significances of Thiamine Deficiency
The earliest symptoms of thiamine deficiency include constipation, appetite suppression, and nausea. Progressive deficiency will lead to mental depression, peripheral neuropathy and fatigue. Chronic thiamine deficiency leads to more severe neurological symptoms including ataxia, mental confusion and loss of eye coordination (nystagmus). A highly diagnostic physical test of thiamine deficiency is vertical nystagmus. Vertical nystagmus is characterized by spontaneous upbeating or downbeating of the eyeball. There are numerous causes or horizontal nystagmus but vertical is only seen due to the CNS damage associated with thiamine deficiency or with phencyclidine (PCP) intoxication. Additional clinical symptoms of prolonged thiamine deficiency are related to cardiovascular and musculature defects.
Dietary thiamine deficiency is known asberiberi, is most often the result of a diet that is carbohydrate rich and thiamine deficient. An additional thiamine deficiency related syndrome is known as Wernicke syndrome which is most often associated with chronic alcohol consumption. This disease is most commonly found in chronic alcoholics due to the fact that alcohol impairs thiamine uptake from the small intestine as well as the fact that these individuals generally have poor dietetic lifestyles. Wernicke syndrome is also referred to as dry beriberi. Prolonged dietary deficiency in thiamine leads to wet beriberi. The wet form of the disease is the result the cardiac involvement in the deficiency. At this stage in the deficiency all four chambers of the heart enlarge due to loss of energy generation and fluid retention resulting in what is called dilated cardiomyopathy. The result of the enlarged chambers is that they can't fill completely resulting in systolic failure. Systole relates to the force associated with cardiac contraction expelling blood to arteries. Blood pumped from the left ventricle enters the aorta and is delivered to the body, whereas blood pumped from the right ventricle is sent to the lungs.
When thiamine deficiency manifests with CNS involvement it is called Korsakoff encephalopathy (or Korsakoff psychosis) and is also commonly referred to as Wernicke-Korsakoff syndrome (WKS). WKS is characterized by acute encephalopathy progressing to chronic impairment of short-term memory. Thiamine supplementation can reverse the symptoms of beriberi and Wernicke syndrome, however, the consequences of severe deficiency (WKS) are irreversible. The confabulation of Korsakoff psychosis is due to destruction of the mammillary bodies in the brain. The mammillary bodies are composed of two small round structures at the underside of the brain that are part of the limbic system, specifically they are part of thePapez circuit. This circuit is also called the hippocampal-mammillo-thalamo-cortical pathway. The consequence of destruction of the mammillary bodies is retrograde amnesia.
Persons afflicted with an inherited form of Wernicke-Korsakoff syndrome appear to have an inborn error of metabolism that is clinically important only when the diet is inadequate in thiamine. These individuals were thought to harbor an abnormality in the enzyme, transketolase. Although a variant transketolase enzyme has been proposed to be associated with Wernicke-Korsakoff syndrome, no mutations have been found in the gene (symbol: TKT) encoding this enzyme when cloned from patients exhibiting the syndrome.
It has been speculated that the protein encoded by a transketolase-related gene (transketolase-like 1: TKTL1) may be involved in the inherited propensity for the development of WKS. However, the TKTL1 encoded protein lacks 38 amino acids, compared to the TKT protein, in the TPP-binding region. All TPP-dependent enzymes contain a highly similar TPP-binding domain and its lack in the TKTL1 protein strongly suggests that it is unlikely that TKTL1 is a TPP-dependent protein capable of catalyzing the transketolase reaction. Indeed, recent evidence has confirmed that the TKTL1 protein does not catalyze the transketolase reaction of the PPP. Intense interest in the TKTL1 gene, and its encoded protein, was stimulated because it was shown that the level of TKTL1 expression correlated with poor patient outcomes and metastasis in many solid tumors. In addition, specific inhibition of TKTL1 mRNA has been shown to inhibit cancer cell proliferation in functional studies.
A Wernicke-like encephalopathy is associated with mutations in one of the thiamine transporter genes. As indicated above, cellular uptake of thiamine occurs via the transporter encoded by the SLC19A3 gene. Mutations in the SLC19A3 gene have been linked to an inherited Wernicke-like disorder.
Riboflavin
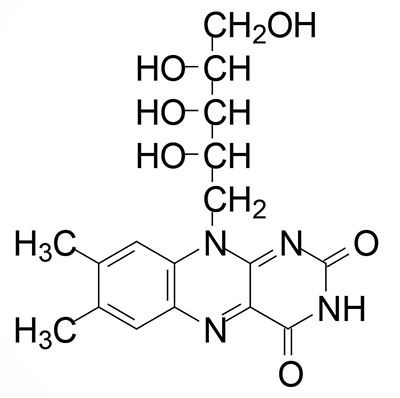
Riboflavin is also known as vitamin B2. Riboflavin is the precursor for the coenzymes, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). Dietary riboflavin is absorbed from the small intestine through the action of the solute carrier family member transporter encoded by the SLC52A3 gene. Cellular uptake of riboflavin occurs through the actions of the SLC transporters encoded by the SLC52A1 and SLC52A2 genes. The SLC52A2 gene is highly expressed in the brain and mutations in this gene result in the autosomal recessive progressive neurological disorder known as Brown-Vialetto-Van Laere syndrome 2.
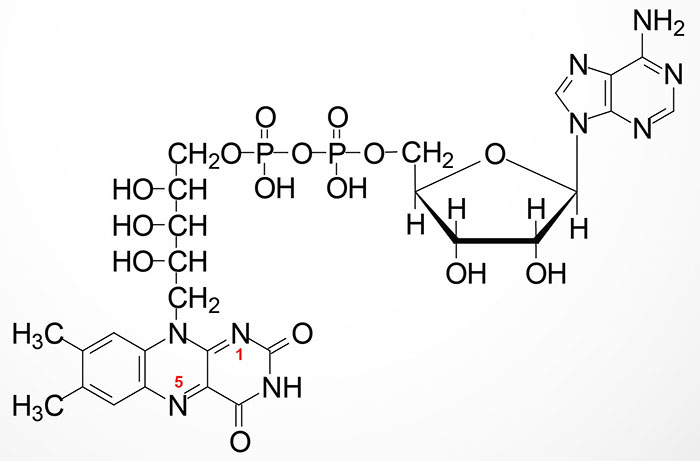
FMN is synthesized from riboflavin via the ATP-dependent enzyme riboflavin kinase (RFK). RFK introduces a phosphate group onto the terminal hydroxyl of riboflavin. The RFK gene is located on chromosome 9q21.13 and is composed of 4 exons that encode a 155 amino acid protein. FMN is then converted to FAD via the attachment of AMP (derived from ATP) though the action of flavin adenine dinucleotide synthetase 1 which is encoded by the FLAD1 gene. The FLAD1 gene is located on chromosome 1q21.3 and is composed of 7 exons that generate four alternatively spliced mRNAs each of which encode distinct isoforms of the enzyme.
The enzymes that require FMN or FAD as cofactors are termed flavoproteins. Several flavoproteins also contain metal ions and are termed metalloflavoproteins. Both classes of enzyme are involved in a wide range of red-ox reactions and includes the same critical thiamine-dependent enzymes described above, the pyruvate dehydrogenase complex (PDHc), 2-oxoglutarate (α-ketoglutarate) dehydrogenase (OGDH), and branched-chain α-ketoacid dehydrogenase (BCKD). Additional important metabolic regulatory enzymes that require flavin as a co-factor include, succinate dehydrogenase (TCA cycle and complex II of oxidative phosphorylation), glycerol-3-phosphate dehydrogenase (involved in the glycerol phosphate shuttle and triglyceride synthesis), and xanthine oxidase involved in purine nucleotide catabolism. During the course of the enzymatic reactions involving the flavoproteins the reduced forms of FMN and FAD are formed, FMNH2 and FADH2, respectively. The hydrogens of FADH2 are on nitrogens 1 and 5 as indicated in the Figure. The normal daily requirement for riboflavin is 1.2–1.7 mg/day for normal adults.
Dietary Sources of Riboflavin
Riboflavin is found in dairy products, lean meats, poultry, fish, grains, broccoli, turnip greens, asparagus, spinach, and enriched food products.
| Food source | Riboflavin content (mg) |
| Beef liver, 100 gm cooked | 4.14 |
| Mackerel, 100 gm canned | 0.54 |
| Pork, loin, 85 gm cooked | 0.24 |
| Hamburger, lean, 100 gm | 0.21 |
| Chicken, dark, 85 gm cooked | 0.19 |
| Steamed clams, 100 gm | 0.43 |
| Yogurt, low-fat, 130 gm | 0.37 |
| Egg, cooked | 0.25 |
| Cheese, cottage, 65 gm | 0.21 |
| Milk, nonfat, 130 gm | 0.34 |
| Pasta, 130 gm cooked | 0.23 |
| Bagel, plain | 0.22 |
| Spinach, 65 gm cooked | 0.16 |
| Wheat germ, raw, 30 ml | 0.12 |
| Soy nuts, 65 gm | 0.65 |
| Almonds, 65 gm | 0.78 |
Clinical Significances of Flavin Deficiency
Riboflavin deficiencies are rare in the United States due to the presence of adequate amounts of the vitamin in eggs, milk, meat and cereals. Riboflavin deficiency is often seen in chronic alcoholics due to their poor dietetic habits.
Symptoms associated with riboflavin deficiency include itching and burning eyes, angular stomatitis and cheilosis (cracks and sores in the mouth and lips), bloodshot eyes, glossitis (inflammation of the tongue leading to purplish discoloration), seborrhea (dandruff, flaking skin on scalp and face), trembling, sluggishness, and photophobia (excessive light sensitivity). Riboflavin decomposes when exposed to visible light. This characteristic can lead to riboflavin deficiencies in newborns treated for hyperbilirubinemia by phototherapy requiring dietary supplementation in these infants.
Niacin
Niacin (nicotinic acid) is also known as vitamin B3. Both nicotinic acid (NA) and nicotinamide (NAM) can serve as the dietary source of the co-factor forms of vitamin B3, nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP).

With respect to niacin the designation NAD (and NADP) refers to the chemical backbone of the active form of the vitamin-derived cofactor. The designations, NAD+ and NADH, as well as NADP+ and NADPH, refer to the oxidized and reduced forms of the cofactors, respectively.
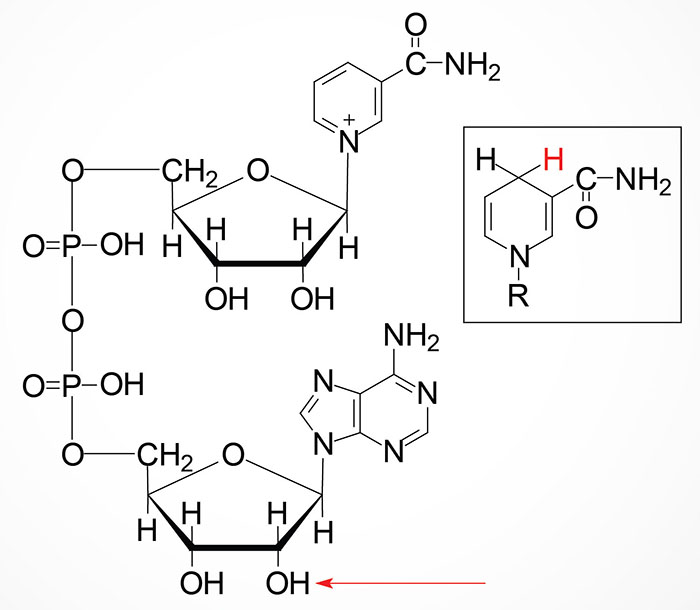
Nicotinamide (NAM) can also be obtained in the diet from the consumption of NAD and NADP, both of which are hydrolyzed to nicotinamide within the lumen of the small intestines. The nicotinamide is then absorbed and delivered to the blood. Nicotinamide can also be hydrolyzed to nicotinic acid (NA) in the lumen of the small intestines and then absorbed. Intestinal uptake of nicotinic acid and nicotinamide is the function of the solute carrier family transporter encoded by the SLC22A13 gene. The SLC22A13 encoded transporter is also involved in high affinity nicotinic acid exchange in the kidneys.
Dietary Source of Niacin
Niacin is not a true vitamin in the strictest definition since NAD can be derived from the amino acid tryptophan. However, the ability to utilize tryptophan for NAD synthesis is inefficient given that approximately 60 mg of tryptophan are required to synthesize 1 mg of NAD. Also, synthesis of NAD from tryptophan requires vitamins B1, B2 and B6 which would be limiting in themselves on a marginal diet. The recommended daily requirement for niacin is 13–19 niacin equivalents (NE) per day for a normal adult. One NE is equivalent to 1 mg of free niacin.
Niacin is found in liver, meat, peanuts and other nuts, and whole grains. In addition, foods that are rich in protein, with exception of tryptophan-poor grains, can satisfy some of the demand for niacin.
| Food source | Niacin content (mg) |
| Beef liver, 100 gm cooked | 14.4 |
| Chicken, white meat, cooked | 13.4 |
| Tuna, canned in water, 85 gm | 11.8 |
| Salmon, 100 gm cooked | 8.0 |
| Ground beef, 100 gm cooked | 5.3 |
| Peanuts, 65 gm | 10.5 |
| Almonds, 65 gm | 1.4 |
| Potato, baked with skin | 3.3 |
| Mushrooms, raw, 65 gm | 1.7 |
| Barley, 65 gm cooked | 1.6 |
| Corn, yellow, 65 gm | 1.3 |
| Lentils, 65 gm cooked | 1.4 |
| Sweet potatoes, 65 gm cooked | 1.2 |
| Carrot, raw, medium | 0.7 |
| Peach, raw, medium | 0.9 |
| Mango, 1 medium | 1.5 |
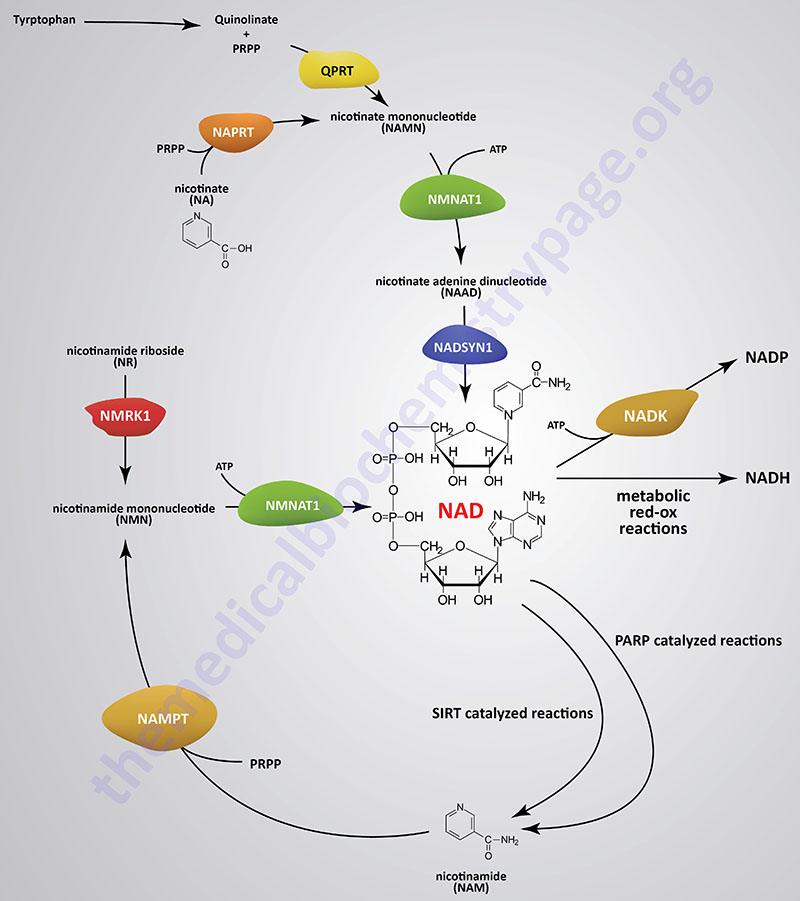
Synthesis and Salvage of NAD+
With the exception of neurons, human cells cannot transport NAD+ into the cell, therefore, they must synthesize it eitherde novo or through the pathway involving the amino acid tryptophan. However, due to the critical need for NAD, most cells recycle the cofactor via salvage pathways as opposed tode novo synthesis. The salvage pathways for NAD synthesis take place in both the nucleus and the mitochondria.
Formation of NAD+ from nicotinamide (NAM) occurs in a two-step process and its formation from nicotinic acid occurs in a three-step process. This latter pathway of NAD+ synthesis is referred to as the Preiss-Handler pathway. Both reaction pathways require the activated form of ribose, 5-phosphoribosyl 1-pyrophosphate (PRPP). PRPP is the same activated ribose required for nucleotide synthesis. Nicotinamide is converted to nicotinamide mononucleotide (NMN) in what is a salvage pathway utilizing nicotinamide phosphoribosyltransferase. Nicotinamide phosphoribosyltransferase is encoded by the NAMPT gene. The NAMPT gene is located on chromosome 7q22.3 and is composed of 11 exons that encode a 491 amino acid precursor protein.
Interestingly there was an activity that was identified at high levels in white adipose tissue that was purported to have insulin mimetic effects. This purported activity was originally called visfatin. The protein identified as visfatin was subsequently discovered to be encoded by the NAMPT gene. The level of NAMPT activity changes under different dietary states such as nutrient status, stress, and during exercise. Of clinical significance is the fact that NAMPT levels are reduced in obesity decreasing its role in stress responses. The NAMPT protein is found both intracellularly (identified as iNAMPT) and extracellularly (identified as eNAMPT). Among numerous functions, the activity of NAMPT is required for the regulation NAD+-dependent deacetylases of the sirtuin (SIRT) family. SIRT enzymes are critical in the regulation of gene expression through their ability to deacetylate histone proteins, thereby altering chromatin structure.
Following the synthesis of NMN, it is then converted to NAD+ through the action of the nicotinamide nucleotide adenylyltransferase (NMNAT) enzymes and ATP. Humans express three NMNAT genes identified as NMNAT1, NMNAT2, and NMNAT3.
The NMNAT1 gene is located on chromosome 1p36.22 and is composed of 9 exons that generate three alternatively spliced mRNAs that collectively encode two distinct protein isoforms. The NMNAT1 encoded proteins are localized to the nucleus.
The NMNAT2 gene is located on chromosome 1q25.3 and is composed of 13 exons that generate two alternatively spliced mRNAs both of which encode distinct protein isoforms. Expression of the NMNAT2 gene is predominantly localized to the brain. The NMNAT2 encoded proteins are found in the cytosol and in the Golgi.
The NMNAT3 gene is located on chromosome 3q23 and is composed of 14 exons that generate seven alternatively spliced mRNAs collectively encode six different protein isoforms. The NMNAT3 encoded proteins are localized to the mitochondria.
The distinct localization of the various NMNAT enzymes indicates that there are likely to be distinct pools of NAD+ in the different subcellular organelles and that these pools are independently regulated through the activity of a given NMNAT.
Nicotinic acid (NA) is converted to nicotinate mononucleotide (NAMN) via the action of nicotinate phosphoribosyltransferase encoded by the NAPRT gene. The NAPRT gene is located on chromosome 8q24.3 and is composed of 12 exons that generate four alternatively spliced mRNAs each encoding distinct isoforms of the enzyme. NAMN is then converted to nicotinate adenine dinucleotide (NAAD) by the action of the NMNAT enzymes. NAAD is then converted to NAD+ via the action of NAD synthetase (also called glutamine-dependent NAD synthetase) which is encoded by the NADSYN1 gene. The NADSYN1 gene is located on chromosome 11q13.4 and is composed of 21 exons that encode a 706 amino acid protein.
NAD+ can be converted to NADP+ through the action of cytoplasmic NAD kinase (NADK) or mitochondrial NAD kinase 2 (NADK2). The NADK gene is located on chromosome 1p36.33 and is composed of 19 exons that generate six alternatively spliced mRNAs that collectively generate four distinct isoforms of the enzyme. The NADK2 gene is located on chromosome 5p13.2 and is composed of 15 exons that generate four alternatively spliced mRNAs, that collectively encode three distinct isoforms of the enzyme. Expression of the NADK2 gene is highest in the liver, being 5-6 fold higher there than in any other tissue.
Mitochondrial NAD+
Whereas the localization of the NMNAT3 encoded protein is in the mitochondria, little, if any, synthesis or salvage of NAD+ has been shown to take place within the mitochondria itself. Given that ~70% of the total pool of NAD+ is found in the mitochondria, where it functions in the TCA cycle and in electron transport through complex 1 of the oxidative phosphorylation pathway, its import into this organelle is critical.
Only recently has the mitochondrial NAD+ import transporter been identified. The protein encoded by the SLC25A51 gene is localized to the inner mitochondrial membrane and is required for transport of NAD+ into the matrix of the mitochondria. The SLC25 family of transporters is known as the mitochondrial carrier family. The SLC25A51 encoded protein is also known as mitochondrial carrier triple repeat protein 1, MCART1.
In experiments in cells where the function of SLC25A51 has been ablated, the level of mitochondrial TCA cycle activity is greatly reduced, the level of mitochondrial respiration and electron transport through complex I is greatly reduced, and the levels of mitochondrial NAD+ and NADH are also significantly reduced. These results indicate a critical role for the SLC25A51 encoded transporter in mitochondrial import of NAD+.
Biological Functions of NAD+ and NADP+
NAD+ functions as a cofactor for numerous dehydrogenases including the same critical thiamine-dependent enzymes described above [the pyruvate dehydrogenase complex (PDHc), 2-oxoglutarate (α-ketoglutarate) dehydrogenase, and branched-chain α-ketoacid dehydrogenase (BCKD)] as well as lactate dehydrogenase and malate dehydrogenase, for example. NADPH serves as a cofactor for numerous reductive biosynthetic processes including fatty acid synthesis, cholesterol biosynthesis, steroid hormone biosynthesis, and bile acid synthesis. In total NAD+/NADP+ is required for over 500 enzymatic reactions and as such plays a critical role in the regulation of almost all major biological processes.
Outside the context of metabolic pathways that utilize NAD+ or NADP+ in red-ox reactions, NAD+ is required for a wide array of biological processes. There are two major cell signaling pathways that utilize NAD+ as a co-substrate. The sirtuin proteins and the poly-ADP-ribose polymerases (PARP) are all NAD+-dependent enzymes that function in processes that include mitochondrial metabolism, inflammation, meiosis, autophagy, apoptosis, transcriptional regulation, RNA biogenesis and processing, DNA damage repair, chromatin remodeling, the unfolded protein response (UPR), and circadian rhythms. The major function of the SIRT proteins is the NAD+-dependent deacetylation of lysine residues in target proteins, particularly histone proteins. During the course of the deacetylation reaction NAM is released and salvaged for synthesis of NAD.
The PARP family of enzymes is encoded by 17 different genes in humans. This family of NAD+-dependent enzymes includes both poly-ADP-ribose polymerases (encoded by the PARP1, 2, and 5 genes) and mono-ADP-ribose transferases (encoded by the PARP3, 4, 6, 10, and 14–16 genes). The reaction catalyzed by PARP enzymes is the cleavage of NAD+ with the subsequent transfer of ADP-ribose to aspartic acid, glutamic acid, and lysine residues on target proteins with the release of NAM. In addition to the PARP family of ADP-ribosylating enzymes, the SIRT4 and SIRT6 encoded enzymes and the arginine-specific ecto-enzymes (ARTC) also possess ADP-ribosylating activity. The process of ADP-ribosylation is a reversible post-translational modification and it's presence in a protein is recognized by specific binding proteins termed ADP-ribose "readers".
NAD+ Degradation
NAD+ is degraded in cells primarily via the actions of two related glycohydrolases. These consumption reactions are distinct from the conversion of NAD+ to NADH or to NADP or to the release of NAM in the context of the use of NAD+ by SIRT or PARP enzymes. The two glycohydrolases are encoded by the CD38 and BST1 (bone marrow stromal cell antigen 1) genes. Both of these enzymes convert NAD+ to nicotinamide (NAM) and adenosine diphosphoribose (ADPR). The NAM is then rapidly converted back to NAD through the salvage pathways described above. The CD38 gene is ubiquitously expressed and the encoded enzyme is the major NAD degrading enzyme in mammals. Expression of the BST1 gene is restricted to lymphoid cells and cells of the gut. As humans age the level of CD38 expression increases leading to a concurrent decline in cellular NAD+ levels. Another NAD+ cleaving enzyme is encoded by the SARM1 (sterile alpha and TIR motif containing 1) gene. Expression of SARM1 is enriched in neural tissues.
Clinical Significance of Niacin
A diet deficient in niacin (as well as tryptophan) leads to glossitis of the tongue (inflammation of the tongue leading to purplish discoloration), dermatitis, weight loss, diarrhea, depression and dementia. The severe symptoms,depression,dermatitis anddiarrhea (referred to as the "3-D's"), are associated with the condition known aspellagra. Several physiological conditions (e.g. Hartnup disorder and malignant carcinoid syndrome) as well as certain drug therapies (e.g. isoniazid) can lead to niacin deficiency. In Hartnup disorder, tryptophan absorption is impaired and in malignant carcinoid syndrome tryptophan metabolism is altered resulting in excess serotonin synthesis. Isoniazid (the hydrazide derivative of isonicotinic acid) was, at one time, a primary drug for chemotherapeutic treatment of tuberculosis.
Nicotinic acid (but not nicotinamide) when administered in pharmacological doses of 2–4 g/day lowers plasma cholesterol levels and has been shown to be a useful therapeutic for hypercholesterolemia. These effects of nicotinic acid are exerted by it binding to and activating the hydroxycarboxylic acid receptor 2 (HCA2) which was originally identified as GPR109A. The major action of nicotinic acid in this capacity is a reduction in fatty acid mobilization from adipose tissue. The HCA2 receptor activates an associated Gi-type G-protein which inhibits the production of cAMP, thereby reducing the PKA-mediated phosphorylation and activation of hormone-sensitive lipase, HSL.
Although nicotinic acid therapy lowers blood cholesterol it also causes a depletion of glycogen stores and fat reserves in skeletal and cardiac muscle. Additionally, there is an elevation in blood glucose and uric acid production. For these reasons nicotinic acid therapy is not recommended for diabetics or persons who suffer from gout. One of the side-effects of nicotinic acid therapy, that reduces patient compliance with its use, is cutaneous vasodilation resulting in a burning flush in the face and upper body. This negative effect of nicotinic acid is the result of the activation of HCA2 on macrophages.
Pantothenic Acid
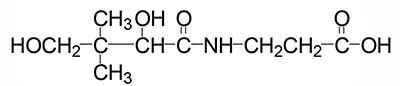
Pantothenic acid is also known as vitamin B5. In humans pantothenic acid is synthesized by colonic bacteria from β-alanine and pantoic acid. The pantothenate is then absorbed from the intestine via the action of a Na+-dependent transporter that is also responsible for biotin absorption from the gut. This vitamin transporter is encoded by the SLC5A6 gene.
Biosynthesis of Coenzyme A (CoASH)
In the synthesis of coenzyme A (abbreviated CoA or CoASH) from pantothenate there are five reaction steps. Pantothenate is phosphorylated on the hydroxyl group via the action of the pantothenate kinases forming 4′-phosphopantethenate. Humans express four pantothenate kinase genes identified as PANK1–PANK4.
The PANK1 gene is regulated by the tumor suppressor, p53. In addition, an intronic miRNA gene resides within the PANK1 gene. The PANK1 gene is located on chromosome 10q23.31 and is composed of 12 exons that generate three alternatively spliced mRNAs each of which encode unique isoforms of the enzyme. Highest levels of PANK1 expression are in the liver and kidney.
The PANK2 enzyme is the only one of the four enzymes to be localized to the mitochondria. The PANK2 gene is located on chromosome 20p13 and is composed of 10 exons that generate seven alternatively spliced mRNAs that collectively encode five distinct protein isoforms. Transcription of one of the PNAK2 mRNAs initiates from a different start site downstream of the major transcriptional start site and translation of the encoded protein begins from a non-AUG codon.
Expression of the PANK3 gene is expressed at highest levels in the intestinal tract. The PANK3 gene is located on chromosome 5q34 and is composed of 7 exons that encode a 370 amino acid protein.
The PANK4 gene is expressed at highest levels in skeletal muscle. The PANK4 gene is located on chromosome 1p36.32 and is composed of 20 exons that encode a 781 amino acid protein. A mutation in intron 4 of the PANK4 gene is associated with autosomal dominant congenital posterior cataract demonstrating a role for the encoded protein in the regulation of lens cell proliferation, lens epithelial cell apoptosis, lens crystallin formation, and fiber cell functions.
The reactive sulfhydryl group is added to the carboxylic acid end of 4′-phosphopantothenate from cysteine via the action of phosphopantothenoylcysteine synthetase which is encoded by the PPCS gene. The PPCS gene is located on chromosome 1p34.2 and is composed of 5 exons that generate eight alternatively spliced mRNAs that collectively encode four distinct isoforms of the enzyme. The product of the PPCS reaction is 4′-phospho-N-pantothenoylcysteine (PPC).
PPC is then decarboxylated to 4′-phosphopantetheine via the action of phosphopantothenoylcysteine decarboxylase (PPCDC). The PPCDC gene is located on chromosome 15q24.2 and is composed of 10 exons that generate six alternatively spliced mRNAs encoding five distinct protein isoforms.
4′-Phosphopantetheine is then converted to coenzyme A via the action of the bifunctional enzyme, coenzyme A synthase encoded by the COASY gene. The phosphopantetheine adenylyltransferase domain of COASY utilizes ATP to catalyze the conversion of 4′-phosphopantetheine into dephospho-coenzyme A. The dephospho-CoA kinase domain of COASY catalyzes the final step in CoA synthesis by adding the phosphate from ATP to the 2′-hydroxyl of the ribose from the adenine added in the previous reaction. The COASY gene is located on chromosome 17q21.2 and is composed of 9 exons that generate three alternatively spliced mRNAs, two of which encode the same enzyme isoform.
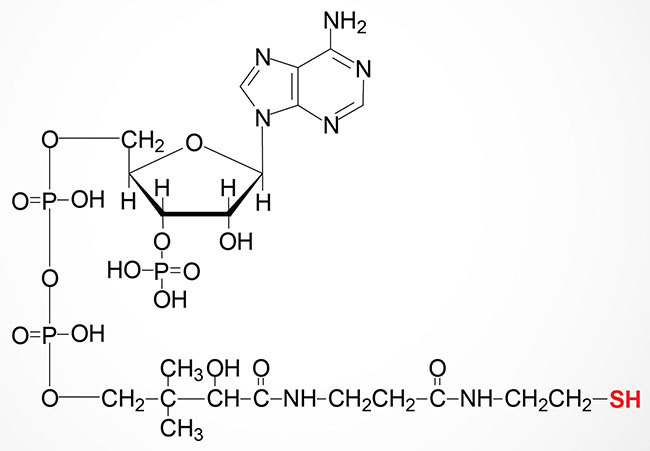
In addition to its role in the synthesis of coenzyme A, pantothenic acid serves as a component of the acyl carrier protein (ACP) domain of fatty acid synthase, FAS. Pantothenate is, therefore, required for the metabolism of carbohydrate via the TCA cycle and all fats and proteins. At least 70 enzymes have been identified as requiring CoA or ACP derivatives for their function including the same critical thiamine-dependent enzymes described above, the pyruvate dehydrogenase complex (PDHc), 2-oxoglutarate (α-ketoglutarate) dehydrogenase (OGDH), and branched-chain α-ketoacid dehydrogenase (BCKD).
Dietary Sources of Pantothenic Acid
| Food source | Vitamin B5 content (mg) |
| Beef liver, 100 gm | 5.3 |
| Poultry, dark meat, 100 gm | 1.3 |
| Poultry, white meat, 100 gm | 1.0 |
| Salmon, 100 gm cooked | 1.4 |
| Low fat yogurt, 130 gm, | 1.5 |
| Milk, nonfat, 130 gm | 0.8 |
| Bleu cheese, 28 gm | 0.49 |
| Cottage cheese, 65 gm | 0.27 |
| Corn, cooked, 65 gm | 0.72 |
| Potato, baked, one | 0.7 |
| Sweet potato, 65 gm | 0.68 |
| Broccoli, boiled, 65 gm | 0.4 |
| Wheat germ, raw, 32 gm | 1.2 |
| Mushrooms, cooked, 65 gm | 0.84 |
| Peanuts, 65 gm | 0.9 |
| Avocado half | 1.0 |
| Sunflower seeds, 32 gm | 2.3 |
| Dates, 10 | 0.65 |
| Papaya, 65 gm | 0.33 |
| Strawberries, 130 gm | 0.25 |
| Orange juice, 235 ml | 0.24 |
Deficiency of pantothenic acid is extremely rare due to its widespread distribution in whole grain cereals, legumes and meat. Symptoms specific to pantothenate deficiency are difficult to assess since they are subtle and resemble those of other B vitamin deficiencies. These symptoms include painful and burning feet, skin abnormalities, retarded growth, dizzy spells, digestive disturbances, vomiting, restlessness, stomach stress, and muscle cramps.
Vitamin B6
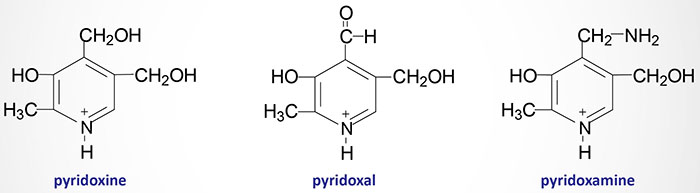
Pyridoxal, pyridoxamine and pyridoxine are collectively known as vitamin B6. All three compounds are efficiently converted to the biologically active form of vitamin B6, pyridoxal phosphate (PLP). This conversion is catalyzed by the ATP requiring enzyme, pyridoxal kinase. Pyridoxal kinase requires zinc for full activity thus making it a metalloenzyme. Pyridoxal kinase is encoded by the PDXK gene which is located on chromosome 21q22.3 and is composed of 19 exons that generate two alternatively spliced mRNAs encoding isoform 1 (312 amino acids) and isoform 2 (272 amino acids).
Any PLP consumed in the diet is acted upon by intestinal alkaline phosphatases that remove the phosphate. All three forms of vitamin B6 are then passively absorbed by intestinal enterocytes of the jejunum. Within intestinal enterocytes the three forms are re-phosphorylated to PLP and then delivered to the blood.
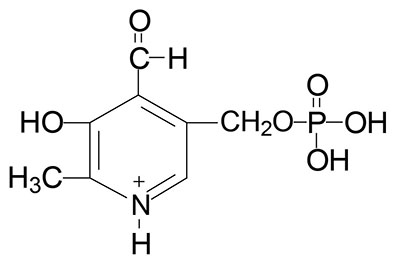
Vitamin B6 Requiring Reactions
Pyridoxal phosphate (PLP) functions as a cofactor in all of the enzymes that carry out the transamination (aminotransferase) reactions required for the synthesis and catabolism of the amino acids. PLP is a cofactor for the synthesis of several neurotransmitters including serotonin, the catecholamines dopamine, norepinephrine, and epinephrine [critical enzyme is aromatic L-amino acid decarboxylase (AADC) which is encoded by the DDC gene and is more commonly referred to as DOPA decarboxylase], and γ-aminobutyric acid, GABA (critical enzyme is glutamic acid decarboxylase, GAD). PLP is required for the synthesis of heme via the enzyme catalyzing the initial and regulated step in this pathway, δ-aminolevulinic acid synthase (ALAS). PLP is a cofactor for two enzymes involved in methionine and cysteine metabolism, cystathionine β-synthase (CBS) and cystathionase (cystathionine γ-lyase). As indicated above, PLP is also required for the conversion of tryptophan to nicotinamide adenine dinucleotide (NAD+). PLP is also required for glycogen homeostasis as a cofactor for glycogen phosphorylase. This latter reaction is the only PLP-dependent process that is not associated with metabolism of nitrogen containing compounds (principally amino acids).
Dietary Sources of Vitamin B6
The requirement for vitamin B6 in the diet is proportional to the level of protein consumption ranging from 1.4–2.0 mg/day for a normal adult. During pregnancy and lactation the requirement for vitamin B6 increases approximately 0.6 mg/day.
| Food source | Vitamin B6 content (mg) |
| Beef liver, 100 gm | 1.4 |
| Turkey, light meat, 100 gm | 0.5 |
| Chicken, light meat, 100 gm | 0.63 |
| Salmon, 100 gm cooked | 0.65 |
| Halibut, baked, 100 gm | 0.4 |
| Potatoes, 130 gm | 0.48 |
| Sweet potatoes, 65 gm | 0.3 |
| Oatmeal, 130 gm cooked | 0.74 |
| Rice, brown, cooked, 130 gm | 0.28 |
| Brussels sprouts, 65 gm | 0.23 |
| Lentils, 65 gm cooked | 0.18 |
| Carrots, 65 gm cooked | 0.18 |
| Peanuts, 65 gm | 0.18 |
| Sunflower seeds, 32 gm | 0.26 |
| Avocado, 1 Haas | 0.48 |
| Mango | 0.28 |
| Watermelon, 130 gm | 0.22 |
| Cantaloupe, 130 gm | 0.18 |
| Prunes, 10 dried | 0.22 |
| Blackstrap molasses, 30 ml | 0.29 |
Vitamin B6 Deficiency and Disease
Deficiencies of vitamin B6 are rare and usually are related to an overall deficiency of all the B-complex vitamins. Like the role of chronic alcohol consumption and an associated poor diet leading to thiamine deficiency, alcoholism is the leading cause of deficiency in B6.
Isoniazid (see niacin deficiencies above) and penicillamine (used to treat rheumatoid arthritis and cystinurias) are two drugs that complex with pyridoxal and PLP resulting in a deficiency in this vitamin.
Deficiencies in pyridoxal kinase result in reduced synthesis of PLP and are associated with seizure disorders related to a reduction in the synthesis of GABA.
Due to its role in heme biosynthesis, deficiency in vitamin B6 can result in microcytic hypochromic anemias that are similar to those caused by iron deficiency or as a result of heavy metal (e.g. lead) poisoning.
Two additional critical enzymes requiring PLP are cystathionine β-synthase (CBS) and cystathionine γ-lyase (cystathionase) which are involved in the metabolism of methionine to cysteine. Due to the role of B6 in this latter reaction, deficiencies in the vitamin can lead to homocysteinemia/uria due to a resultant blockade in the CBS reaction (see the Amino Acid Metabolism page for discussion of this effect).
Other symptoms that may appear with deficiency in vitamin B6 include nervousness, insomnia, skin eruptions, loss of muscular control, anemia, mouth disorders, muscular weakness, dermatitis, arm and leg cramps, loss of hair, slow learning, and water retention.
Differential Diagnosis: Several Causes of Microcytic Anemia
| Deficiency/Defect | Characteristics |
| B6 deficiency | PLP is required for the rate-limiting enzyme in heme biosynthesis: δ-aminolevulinic acid synthase (ALAS); deficiency results in loss of protoporphyrin IX synthesis, therefore, there will be a significantreduction in measurable ALAS product (δ-aminolevulinic acid, δ-ALA) and protoporphyrin in these patients; loss of heme production leads to hypochromic microcytic anemia; lack of protoporphyrin results in iron deposits on mitochondria in bone marrow erythroblasts resulting in the formation ofringed sideroblasts; loss of iron incorporation into protoporphyrin IX leads toincreased serum and intracellular iron concentration; increase in intracellular iron results inincreased translation of ferritin as a means to prevent iron toxicity |
| Iron deficiency | iron deficiency is the leading cause of microcytic anemia; loss of iron results in reduced production of heme, thus, the result is a hypochromic microcytic anemia; lack of heme production results in loss of feed-back inhibition of ALAS, therefore these patients will have an associatedincrease in measurable protoporphyrin; loss of iron intake meansreduced iron in the serum and reduced intracellular iron, the latter resulting inreduced ferritin translation; loss of iron for incorporation into protoporphyrin IX results in spontaneous, non-enzymatic incorporation of Zn2+ formingZn-protoporphyrin (ZPP), ZPP causes erythrocytes to fluoresce under ultraviolet illumination and is the basis of the ZPP test for iron deficiency or lead poisoning |
| Heavy metal poisoning | heavy metals, such as lead, inhibit several enzymes of heme biosynthesis and metabolism with the most significant toxic effects resulting from inhibition of ferrochelatase, the enzyme that incorporates iron into protoporphyrin IX generating heme; similar to B6 deficiency, lead poisoning leads toincreased intracellular iron in bone marrow erythroblasts causing the formation ofringed sideroblasts; because there is no heme, the ALAS reaction is not inhibited, as in the case of iron deficiency, this results inincreased production of δ-ALA and protoporphyrin; lack of iron incorporation into protoporphyrin results inincreased serum and intracellular iron concentrations, with the latter leading toincreased ferritin synthesis as in the case of iron-deficient anemia; loss of iron for incorporation into protoporphyrin IX results in spontaneous, non-enzymatic incorporation of Zn2+ formingZPP; as in the case of iron-deficient anemia, the ZPP test is diagnostic for lead/heavy metal poisoning |
Biotin
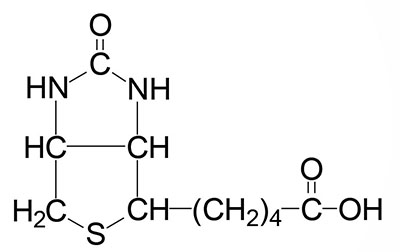
Biotin is the cofactor required of enzymes that are involved in carboxylation, decarboxylation, or transcarboxylation reactions in prokaryotes and eukaryotes. Biotin is sometimes referred to as vitamin H. Biotin, produced by intestinal bacteria, as well as that found in the diet, is bound to lysine residues in protein forming a complex that is called biocytin.
Removal of biotin from biocytin and recycling of biotin from human biotin-dependent enzymes requires the activity of the enzyme biotinidase. The biotinidase gene (gene symbol: BTD) is located on chromosome 3p25.1 and is composed of 11 exons that generate eight alternatively spliced mRNAs that collectively encode four distinct protein isoforms. The clinical significance of defects in the BTD gene are described below.
When released from protein, or consumed in free form, biotin is absorbed from the lumen of the intestines through the action of the Na+-dependent vitamin transporter encoded by the SLC5A6 gene. This is the same transporter involved in pantothenate (vitamin B5) absorption.
In humans, the biotin-requiring enzymes include acetyl-CoA carboxylase (ACC: humans express two distinct ACC genes), pyruvate carboxylase (PC), propionyl-CoA carboxylase (PCC), and 3-methylcrotonyl-CoA carboxylase (3MCC). The critically important biotin-requiring enzymes are ACC, PC, and PCC. All four of these enzymes are referred to asABC enzymes because they require/utilizeATP,Biotin, andCO2.
The biotin-dependent carboxylating enzymes in mammals are multifunctional and contain three distinct enzymatic activities that may be contained in a single protein or in different subunits of the multisubunit enzymes. These three enzymatic activities are the biotin carboxylase (BC), the carboxyltransferase (CT), and the biotin carboxyl carrier protein (BCCP) activities.
Incorporation of Biotin into Biotin-Requiring Enzymes
Incorporation of biotin into the biotin-requiring enzymes involves the activity of holocarboxylase synthetase (also known as protein-biotin ligase) that is encoded by the HLCS gene. The HLCS gene is located on chromosome 21q22.13 and is composed of 20 exons that generate eight alternatively spliced mRNAs, seven of which encode the same 726 amino acid protein.
The reaction of biotin incorporation into the biotin-requiring enzymes occurs in a two-step process that requires ATP. In the first step holocarboxylase synthetase generates a biotin-AMP intermediate and in the second step the activated biotin is transferred to the carboxylase enzyme with release of AMP.
The removal of biotin from lysine residues in proteins in the diet and from the carboxylases in cells, through the action of biotinidase, and then the incorporation of the biotin into the carboxylases through the action of holocarboxylase synthetase is referred to as the biotin cycle.
Biotin-Requiring Enzymes
Acetyl-CoA Carboxylases
The ACC enzymes are cytosolic enzymes with the ACC1 gene encoded protein catalyzing the rate-limiting step of fatty acid synthesis. PC is a mitochondrial enzyme that catalyzes the critical first reaction in the pathway of gluconeogenesis. PCC is a mitochondrial enzyme that catalyzes reactions involved in the metabolism of several amino acids (valine, methionine, isoleucine, and threonine) as well as the oxidation of fatty acids with an odd number of carbon atoms. The role of PCC in the metabolism of these compounds is often referred to as theVOMIT pathway (Valine,Odd-chain fats,Methionine,Isoleucine,Threonine) for memorization purposes. 3MCC is a mitochondrial enzyme that catalyzes the fourth step in the catabolism of leucine.
As discussed in detail in the Fatty Acid, Triglyceride, and Phospholipid Synthesis page, humans express two forms of ACC (ACC1 and ACC2) both of which are multifunctional enzymes possessing all three biotin-dependent carboxylating activities.
Pyruvate Carboxylase
Pyruvate carboxylase is the critical first enzyme in the conversion of pyruvate to glucose in the pathway of gluconeogenesis. Pyruvate carboxylase (PC) is also a multifunctional enzyme containing all three activities in a single polypeptide that form an α4 homotetrameric enzyme. The human PC gene is located on chromosome 11q13.2 and contains 32 exons that generate three alternatively spliced mRNAs. These mRNAs differ in the 5′-untranslated region (5′-UTR) but all three encode the same precursor protein of 1178 amino acids. The highest levels of PC expression are in adipose tissue.
Propionyl-CoA Carboxylase
Propionyl-CoA carboxylase converts the propionyl-CoA derived from the oxidation of valine, methionine, isoleucine, and threonine, as well as fatty acids with odd numbers of carbon acids, into methylmalonyl-CoA. Propionyl-CoA carboxylase functions as a heterododecameric enzyme (subunit composition: α6β6) and the two different subunits are encoded by the PCCA and PCCB genes, respectively. The α-subunit of propionyl-CoA carboxylase contains the three activities of a typical biotin-dependent carboxylase. The BC domain of the α-subunit resides in the N-terminal region and the BCCP domain resides in the C-terminal region. Propionyl-CoA carboxylase has a domain, called the biotin transfer (BT) domain, that is not found in the other biotin-dependent carboxylase family enzymes. The BT domain is found in the middle of the α-subunit and is required for interactions between the α- and β-subunits.
The PCCA gene is located on chromosome 13q32.3 and is composed of 32 exons that generate eleven alternatively spliced mRNAs, each of which encode a unique protein isoform. The PCCB gene is located on chromosome 3q22.3 and is composed of 17 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 539 amino acids (isoform 1) and 559 amino acids (isoform 2).
3-Methylcrotonyl-CoA Carboxylase
The function of 3-methylcrotonyl-CoA carboxylase is to catalyze the carboxylation of 3-methylcrotonyl-CoA to 3-methylglutaconyl-CoA during the catabolism of leucine. The 3-methylcrotonyl-CoA is the product of isovaleryl-CoA dehydrogenase (IVD) which catalyzes the prior reaction in leucine catabolism. 3-methylcrotonyl-CoA carboxylase, 3-Methylcrotonyl-CoA carboxylase (3MCC), like PCC, is also a heterododecameric enzyme (subunit composition: α6β6). Similar to the activities of PCC, the α-subunit possesses the BC and BCCP activities and the β-subunit possesses the CT activity. The α-subunit of 3MCC is encoded by the MCCC1 gene and the β-subunit is encoded by the MCCC2 gene.
The MCCC1 gene is located on chromosome 3q27.1 and is composed of 22 exons that generate three alternatively spliced mRNAs, two of which are known to encode functional protein. MCCC1 isoform 1 is a precursor protein of 725 amino acids and isoform 2 is a precursor protein of 608 amino acids. Mutations in the MCC1 gene are the cause of 3-methylcrotonylglycinuria type I.
The MCCC2 gene is located on chromosome 5q13.2 and is composed of 19 exons that generate two alternatively spliced mRNAs that encode precursor proteins of 563 and 525 amino acids. Mutations in the MCC2 gene are the cause of 3-methylcrotonylglycinuria type II.
Dietary Sources of Biotin
Biotin is found in numerous foods and also is synthesized by intestinal bacteria. Some of the richest sources of biotin are swiss chard, tomatoes, romaine lettuce, and carrots. Additional sources include onions, cabbage, cucumber, cauliflower, mushrooms, peanuts, almonds, walnuts, oat meal, bananas, raspberries, strawberries, soy, egg yolk, and cow and goat milk.
Clinical Significance of Biotin
Given that biotin is synthesized by intestinal bacteria deficiencies of the vitamin are rare. Deficiencies are generally seen only after long antibiotic therapies which deplete the intestinal microbiota or following excessive consumption of raw eggs. The latter is due to the affinity of the egg white protein, avidin, for biotin preventing intestinal absorption of the biotin. An important autosomal recessive inherited disorder that leads to biotin deficiency is biotinidase (BTD) deficiency.
Biotinidase Deficiency
Profound biotinidase deficiency is characterized by mutations in the gene that result in enzyme activity that is less than 10% of the normal. Partial biotinidase deficiency is characterized by enzyme activity that is 10%–30% of normal. Profound biotinidase deficiency occurs with a frequency of 1 in 60,000 live births. Indeed, the frequency is high enough, and the resultant symptoms severe enough, that current neonatal disease testing includes analysis for defects in the activity of this enzyme. The most severe symptoms associated with biotin deficiency and profound biotinidase gene defects are the result of the accumulation of toxic metabolic intermediates. The symptoms of profound biotinidase deficiency include delayed development, seizures, hypotonia, respiratory difficulties, hearing and vision loss, ataxia, skin rashes, and alopecia. Patients with profound biotinidase deficiency are also highly susceptible the fungal infection, candidiasis. Symptoms associated with partial biotinidase deficiency can be similar to those of the profound deficiency form but often these symptoms do not appear as severe except during infections, illnesses, or stress.
Holocarboxylase Synthetase Deficiency
Deficiency in holocarboxylase synthetase is an autosomal recessive disorder that results in multiple carboxylase deficiency. At least 35 different mutations in the HLCS gene have been found associated with holocarboxylase synthetase deficiency. The typical presentation of holocarboxylase synthetase deficiency is seen in the neonatal period with onset occurring within hours to weeks of birth. The classic symptoms of holocarboxylase synthetase deficiency are hypotonia, lethargy, seizures, metabolic acidosis, vomiting, hyperammonemia, developmental delay, skin rash, and alopecia. Standard newborn screening tests include detection of holocarboxylase synthetase deficiency by analysis of blood for the presence of elevated O-(3-hydroxyvaleryl)-L-carnitine. Urine organic acid analysis in holocarboxylase synthetase deficient patients will show elevated levels of lactate, 3-hydroxyisovaleric acid, 3-hydroxypropionic acid, 3-methylcrotonic acid, methylcitric acid, and tiglylglycine. Tigylglycine is an N-acylglycine formed from glycine with an amine hydrogen substituted by a 2-methylbut-2-enoyl (tiglyl) group.
Cobalamin: Vitamin B12
Cobalamin is more commonly known as vitamin B12. Vitamin B12 is composed of a complex tetrapyrrol ring structure (corrin ring) and a cobalt ion in the center. Vitamin B12 (as the hydroxocobalamin form) is synthesized exclusively by microorganisms and is primarily found in the liver of animals bound to protein as methycobalamin or 5′-deoxyadenosylcobalamin. Bioavailable vitamin B12 is also present in cows milk and soybeans. The vitamin must be released from these binding proteins in order to be active and this release occurs in the stomach by the gastric protease, pepsin.
After being released, cobalamin is bound to an endogenous protein called haptocorrin (also known as transcobalamin I, which is encoded by the TCN1 gene). The TCN1 gene is located on chromosome 11q12.1 and is composed of 9 exons that encode a precursor protein of 433 amino acids. Expression of the TCN1 gene highest in the salivary glands. Expression levels in the gallbladder, stomach, and bone marrow are next highest but are 2-3 times lower than that of the salivary glands.
Haptocorrin is a glycoprotein that is produced by oral salivary glands in response to intake of food. The binding of hatocorrin to the cobalamin that is released by the action of pepsin, protects the vitamin while it transits through the stomach into the duodenum. Within the small intestine pancreatic trypsin hydrolyzes the haptocorrin-cobalamin complexes allowing binding of cobalamin to another protein called intrinsic factor, IF. Intrinsic factor is also called gastric intrinsic factor. Intrinsic factor is a heavily glycosylated protein that is homologous to haptocorrin. Intrinsic factor binds cobalamin only within the alkaline pH of the intestines. Haptocorrin and intrinsic factor are two of the three vitamin B12-binding proteins produced in humans.
The third vitamin B12-binding protein is transcobalamin II which is a plasma globulin that is the primary transport protein for vitamin B12 in the blood. Transcobalamin II (often called simply transcobalamin, TC) is encoded by the TCN2 gene. The TCN2 gene is located on chromosome 22q12.2 and is composed of 9 exons that generate two alternatively spliced mRNAs encoding precursor proteins of 427 amino acids (isoform 1) and 400 amino acids (isoform 2).
Intrinsic factor is a 50-kDa glycoprotein produced by the parietal cells of the stomach. Intrinsic factor is encoded by the CBLIF (cobalamin binding intrinsic factor) gene which is also referred to as gastric intrinsic factor (GIF). The CBLIF gene is located on chromosome 11q12.1 and is composed of 9 exons that encode a 417 amino acid precursor protein. Expression of the CBLIF gene is exclusive to parietal cells of the stomach. The secretion of intrinsic factor is stimulated by all three of the mechanisms that lead to gastric acid secretion: histamine, acetylcholine, and gastrin.
Cobalamin, bound to intrinsic factor, can then bind to a receptor complex called cubilin present on the apical membranes of enterocytes in the distal ileum as well as on renal epithelial cells of the nephron in the kidney. The cubilin protein is encoded by the CUBN gene which is located on chromosome 10p13 and is composed of 71 exons that encode a 3623 amino acid precursor protein. Expression of the CUBN gene is highest in the kidney and the small intestine.
Interactions of the intrinsic factor-cobalamin complexes with cubilin requires Ca2+ ions. The cubilin protein is a peripheral membrane protein that lacks both transmembrane and cytoplasmic domains. Internalization of the cubilin bound intrinsic factor-cobalamin complex requires that cubilin be colocalized (anchored) with another transmembrane protein. Two proteins have been shown to fulfill the role of cubilin anchoring proteins. One of these proteins was originally identified in the mutant mouse called amnionless. The human protein is called amnion-associated transmembrane protein and it is encoded by the AMN gene. The other cubilin anchoring protein is a member of the LDL receptor-related protein (LRP) family, specifically LRP2. The AMN protein is required for intestinal uptake of cubilin-intrinsic factor-cobalamin complexes, whereas LRP2 appears to have some role in the uptake of these complexes from the nephron. However, because the disease known as Donnai-Barrow syndrome, which results from inherited mutations in the LRP2 gene, is not associated with vitamin B12 deficiency, the interactions between cubilin and LRP2 is not required for overall vitamin B12 homeostasis. It is suggested that an additional cubilin anchoring protein may yet be identified. Upon intrinsic factor-cobalamin binding to cubilin, the complex is endocytosed. The intrinsic factor in the endocytosed complexes is degraded by lysosomal hydrolases and the cobalamin is released.
Within intestinal enterocytes cobalamin is bound to transcobalamin II (identified as TC-Cbl) for transfer from the apical (lumenal) side to the basolateral (blood) side of the cell. Free cobalamin is released from the intestinal enterocytes to the blood via TC-Cbl interaction with a basolateral membrane transporter that is a member of the ATP-binding cassette family of transporters, specifically the ABCC1 transporter. Within the blood, free cobalamin binds again to transcobalamin II. The transcobalamin II-cobalamin (TCN2-cobalamin) complexes are taken up, from the blood, by all cells via a plasma membrane transcobalamin receptor (TCBLR) encoded by the CD320 gene. The TCN2-cobalamin-TCBLR complex is internalized via endocytosis. Within the lysosome transcobalamin II is degraded and the free cobalamin is released so that it can be used as an enzyme co-factor within the cell. The release of free cobalamin from the lysosome requires the action of the lysosomal membrane protein identified as LMBRD1 (LMBR1 domain containing 1, where LMBR1 refers to the protein identified as limb region 1 protein homolog).
Free within the cytoplasm of the cell cobalamin binds to the protein encoded by the MMACHC gene [methylmalonic aciduria (cobalamin deficiency) cblC type, with homocystinuria]. The MMACHC encoded protein dealkylates both adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl) and also decyanates cyanocobalamin (CN-Cbl). Within the cytosol cobalamin is converted to MeCbl within the context of the methionine synthase reaction as outlined below. Cobalamin is also transported into the mitochondria via the action of the protein encoded by the MMAA gene [methylmalonic aciduria (cobalamin deficiency) cblA type]. Within the mitochondrion cobalamin is converted to AdoCbl which is the co-enzyme form of vitamin B12 required by methylmalonyl-CoA mutase (see below). Conversion of cobalamin to AdoCbl is catalyzed by the protein encoded by the MMAB gene [methylmalonic aciduria (cobalamin deficiency) cblB type]. The MMAB encoded protein synthesizes AdoCbl from cobalamin [the cob(I)yrinic acid a,c-diamide form] in an ATP-dependent reaction. Deficiencies in all of the genes responsible for overall metabolism of cobalamin are associated with various forms of methylmalonic acidemia.
There are only two clinically significant reactions in the body that require vitamin B12 as a co-factor. During the catabolism of fatty acids with an odd number of carbon atoms and the amino acids valine, isoleucine, methionine, and threonine, the resultant propionyl-CoA is converted to succinyl-CoA for oxidation in the TCA cycle. The catabolism of these compounds into propionyl-CoA is often remembered by the mnemonicVOMIT pathway, whereV is for valine,O is for odd number carbon atom fats,M is for methionine,I is for isoleucine, andT is for threonine.
The last enzyme in this pathway, methylmalonyl-CoA mutase, requires vitamin B12 as a cofactor in the conversion of methylmalonyl-CoA to succinyl-CoA which then enters the TCA cycle. The 5′-deoxyadenosine derivative of cobalamin (adenosylcobalamin: AdoCbl) is required for this reaction. Methylmalonyl-CoA mutase is a mitochondrial enzyme and the synthesis of the required AdoCbl occurs within the mitochondria and is catalyzed by the MMAB encoded protein as described earlier.
The second reaction requiring vitamin B12 catalyzes the conversion of homocysteine to methionine and is catalyzed by methionine synthase (also known as homocysteine methyltransferase). This reaction results in the transfer of the methyl group fromN 5-methyltetrahydrofolate (5-methylTHF) to hydroxocobalamin generating tetrahydrofolate (THF) and methylcobalamin (MeCbl) during the overall reaction process.
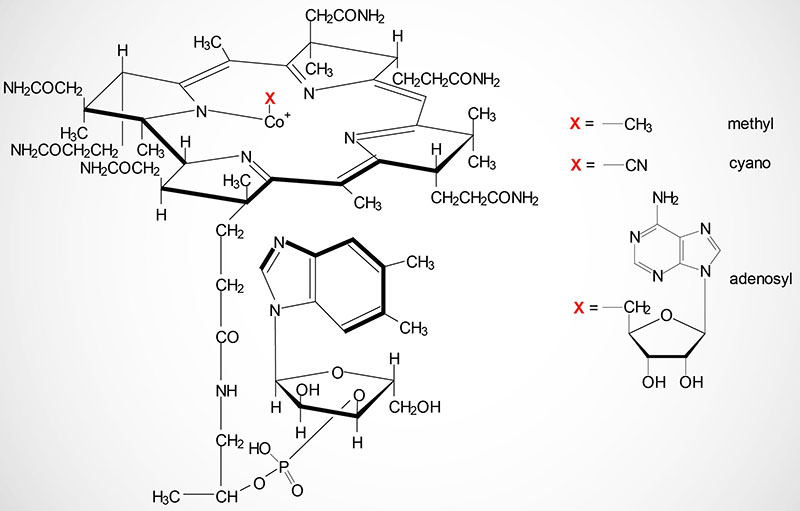
Dietary Source of Vitamin B12
The recommended daily intake (RDA) of vitamin B12 for adults is 2.4 micrograms (μg). Vitamin B12 is found primarily in animal products. Due to the lack of sufficient vitamin B12 in plant foods it is added to breakfast cereals and this serves as a good source of the vitamin for vegetarians. Two plant sources that are useful for obtaining vitamin B12 are alfalfa and comfrey (also written comfry). However, to ensure adequate intake vegans should use a vitamin B12 supplement that contains at least 5–10μg due to the low absorption rate of the vitamin in supplement form.
| Food source | Vitamin B12 content (mcg: μg) |
| Beef liver, 100 gm | 48 |
| Rainbow trout, wild, 85 gm | 5.4 |
| Salmon, 85 gm | 4.9 |
| Beef, 85 gm | 2.4 |
| Tuna, white, 85 gm | 1.0 |
| Yogurt, plain, 130 gm | 1.4 |
| Fortified cereal, 100% RDA | 6.0 |
| Milk, 235 ml | 0.9 |
| Swiss cheese, 30 gm | 0.9 |
| Egg, 1 whole | 0.6 |
Clinical Significance of B12 Deficiency
The liver can store up 3-5 years worth of vitamin B12, hence deficiencies in this vitamin are rare. Pernicious anemia is a megaloblastic anemia resulting from vitamin B12 deficiency that develops as a result of a lack of intrinsic factor being released from stomach parietal cells leading to subsequent malabsorption of the vitamin. The major cause of the loss of intrinsic factor is an autoimmune destruction of the parietal cells that secrete it. Surgical removal of a part of the stomach can also result in loss of intrinsic factor resulting in pernicious anemia. In rare cases individuals inherit mutation in the GIF gene that encodes intrinsic factor resulting in pernicious anemia.
The pernicious anemias result from impaired DNA synthesis due to a block in purine and thymidine biosynthesis. The block in nucleotide biosynthesis is a consequence of the effect of vitamin B12 on folate metabolism. When vitamin B12 is deficient essentially all of the folate becomes trapped as theN 5-methylTHF (5-methylTHF; or simply methylTHF) derivative as a result of the loss of functional methionine synthase (also called homocysteine methyltransferase). This trapping prevents the synthesis of other THF derivatives required for the purine and thymidine nucleotide biosynthesis pathways.
Neurological complications also are associated with vitamin B12 deficiency and result from a progressive demyelination of nerve cells. The demyelination is thought to result from the increase in methylmalonyl-CoA that results from vitamin B12 deficiency that is associated with loss of methylmalonyl-CoA mutase activity. The loss of methylmalonyl-CoA mutase activity with deficiencies in B12 results in an accompanying methylmalonic acidemia. Methylmalonyl-CoA is a competitive inhibitor of malonyl-CoA in fatty acid biosynthesis as well as being able to substitute for malonyl-CoA in any fatty acid biosynthesis that may occur. Since the myelin sheath that protects nerve cells is in continual flux the methylmalonyl-CoA-induced inhibition of fatty acid synthesis results in the eventual destruction of the sheath. The incorporation methylmalonyl-CoA into fatty acid biosynthesis results in branched-chain fatty acids being produced that may severely alter the architecture of the normal membrane structure of nerve cells.
Contributing to the neural degeneration seen in vitamin B12 deficiency is the reduced synthesis of S-adenosylmethione (SAM; also abbreviated AdoMet) due to loss of the methionine synthase catalyzed reaction. The conversion of phosphatidylethanolamine (PE) to phosphatidylcholine (PC) requires the enzyme phosphatidylethanolamineN-methyltransferase (encoded by the PEMT gene) which carries out three successive SAM-dependent methylation reactions. This reaction is a critically important reaction of membrane lipid homeostasis. Therefore, the reduced capacity to carry out the methionine synthase reaction, due to nutritional or disease mediated deficiency of vitamin B12, results in reduced SAM production and as a consequence contributes to the neural degeneration (i.e. depression, peripheral neuropathy) seen in chronic B12 deficiency.
Deficiencies in B12 can also lead to elevations in the level of circulating homocysteine and elevated excretion of the oxidized dimer of homocysteine called homocystine. Elevated levels of homocysteine are known to lead to cardiovascular dysfunction. Due to its high reactivity to proteins, homocysteine is almost always bound to proteins, thus thiolating them leading to their degradation. Homocysteine also binds to albumin and hemoglobin in the blood. Some of the detrimental effects of homocysteine are due to its' binding to lysyl oxidase, an enzyme responsible for proper maturation of the extracellular matrix proteins collagen and elastin. Production of defective collagen and elastin has a negative impact on arteries, bone, and skin and the effects on arteries are believed to be the underlying cause for cardiac dysfunction associated with elevated serum homocysteine. In individuals with homocysteine levels above ≈12μM there is an increased risk of thrombosis and cardiovascular disease. The increased risk for thrombotic episodes, such as deep vein thrombosis (DVT), associated with homocysteinemia is due to homocysteine serving as a contact activation nucleus for activation of the intrinsic coagulation cascade.
Folic Acid

Folic acid is sometimes referred to as vitamin B9. The terms folic acid and folate are sometimes used interchangeably but from a dietary perspective they are distinctly different. The term folate should be used to refer only to the bioactive forms of folic acid, namely dihydrofolate (DHF) and tetrahydrofolate (THF) and their derivatives. Folic acid is a conjugated molecule consisting of a pteridine ring structure linked to para-aminobenzoic acid (PABA) that forms pteroic acid. Pteroic acid is then converted to folic acid through the N-esterification of glutamic acid to the carboxylic acid of the PABA portion of pteroic acid. This latter structure is the form of "folate" present in dietary supplements and when used to fortify manufactured food products. Dietary folates (which are predominately theN 5-methyl-THF form; 5-methyl-THF) are obtained primarily from yeasts and leafy vegetables as well as animal liver. Humans cannot synthesize PABA nor attach glutamate residues to pteroic acid, thus, requiring folic acid or folates (DHF or THF) intake in the diet.
When ingested from natural sources, or when stored in cells (predominantly in the liver and the kidneys), folates exists in a polyglutamate form. Jejunal (small intestine) mucosal cells remove the glutamate residues to the monoglutamate state through the action of the enzyme, folate hydrolase 1 which is encoded by the FOLH1 gene. Folate hydrolase 1 is also commonly called glutamate carboxypeptidase II. The FOLH1 gene is located on chromosome 11p11.12 and is composed of 21 exons that generate six alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Within the intestinal enterocytes, the monoglutamate forms of the folates are less negatively charged (compared to the polyglutamic acids), and are therefore, more capable of being transported across the basolateral membrane (facing the blood) of the jejunal enterocytes and into the bloodstream.
Another enzyme, with similar activity, is called γ-glutamyl hydrolase (also known as conjugase or folate conjugase) and is encoded by the GGH gene. The GGH gene is located on chromosome 8q12.3 and is composed of 10 exons that encode a 318 amino acid precursor protein.
The function of the GGH encoded enzyme is to remove polyglutamates from intracellular stores of the folate derivatives that, as indicated, are predominantly stored in the liver and the kidneys. The GGH encoded enzyme is localized to the lysosomes. The function of the GGH encoded enzyme is to release glutamates from stored folates so that the monoglutamate forms can be excreted into the blood to meet systemic needs.
Folates in the diet are predominantly theN 5-methyltetrahydrofolate (5-methyl-THF) form. Dietary folates are absorbed by jejunal enterocytes primarily via the SLC46A1 encoded transporter which is a proton (H+)-coupled transporter more commonly referred to as the proton-coupled folate transporter, PCFT.
Another intestinal folate transporter, that was originally thought to be the major intestinal folate uptake transporter, was originally identified as the reduced folate carrier (RFC). The RFC protein is encoded by the SLC19A1 gene. Outside the intestine cellular folate uptake, particularly cells in the central nervous system, is carried out by the SLC46A1 encoded transporter. The activity of RFC-mediated folate transport is highest for reduced folates (hence the name) such as 5-methyl-THF with very low affinity for folic acid. The RFC is also utilized in the uptake of the anti-folate drugs such as methotrexate and palatrexate. RFC functions as an antiporter with organic phosphate being transported out of the cell in exchange for folate uptake. Both PCFT and RFC are highly specific for the monoglutamate forms of the folates.
The critical role of the SLC46A1 encoded transporter in intestinal folate absorption is evident in individuals with an inherited form of folate malnutrition (hereditary folate malabsorption, HFM) that results from mutations in the SLC46A1 gene. The SLC46A1 gene is located on chromosome 17q11.2 and is composed of 6 exons that generate two alternatively spliced mRNAs with the major apical membrane-localized protein being the larger isoform 1 (459 amino acids).
Interestingly there are transporters of the multidrug resistance family that can efflux folates from the intestinal enterocyte back into the lumen of the intestines. Both the multidrug resistance-associated protein 2 (MRP2; encoded by the ABCC2 gene) and the breast cancer resistance protein (BCRP; encoded by the ABCG2 gene) are expressed in the apical membranes of intestinal enterocytes. Both transporters have been shown to efflux folates from intestinal enterocytes and, as such, can compete with the activity of PCFT in folate uptake. Another member of the multidrug resistance protein family, multidrug resistance-associated protein 3 (MRP3; encoded by the ABCC3 gene), is expressed in the basolateral membrane of intestinal enterocytes and transports folic acid and 5-methyl-THF into the blood.
Another important protein involved in folate transport in the adult human is the FOLR1 (folate receptor 1) encoded receptor protein. The FOLR1 gene is located on chromosome 11q13.4 and is composed of 7 exons that generate four different mRNAs either through alternative promoter usage or by alternative splicing, all of which encode the same 257 amino acid precursor protein. The FOLR1 encoded protein binds folic acid and the reduced folate derivative (5-methyl-THF) facilitating their transport into cells. The FOLR1 protein exists in soluble form as well as bound to membranes via a GPI-linkage. The FOLR1 gene represents one member of a family of folate receptor genes that are clustered at the 11q13.4 locus. This family includes FOLR1, FOLR2, and FOLR3. The FOLR2 gene is referred to as the fetal folate receptor gene given it was originally identified as being expressed in the placenta. The FOLR2 protein is both secreted and GPI anchored like the FOLR1 protein. The FOLR3 gene is expressed in bone marrow, thymus, and spleen, and its expression is elevated in ovarian and uterine cancers. The FOLR3 protein is exclusively secreted.
When monoglutamate folates are taken up into cells, particularly in the liver and kidney where they can be stored (but also in other cells), they are polyglutamated through the action of the enzyme folylpolyglutamate synthetase. Folylpolyglutamate synthetase is encoded by the FPGS gene which is located on chromosome 9q34.11 and is composed of 17 exons that generate three alternatively spliced mRNAs, each of which encode a different isoform of the enzyme. The activity of folylpolyglutamate synthetase is lowest towards 5-methyl-THF and this prevents this form of the vitamin from being trapped within intestinal enterocytes. Folic acid, which is the form found in fortified foods and supplements is also a poor substrate for folylpolyglutamate synthetase accounting for its efficient uptake from the intestines and release to the blood.
Within cells (principally the liver where it is stored) folic acid found in enriched food and in vitamin supplements is converted first to dihydrofolate (DHF) and then to tetrahydrofolate (THF also H4folate) through the action of dihydrofolate reductase (DHFR), an NADPH-requiring enzyme. The level of DHFR activity in the human liver is relatively low such that high levels of folic acid intake (such as by megadosing vitamins) can lead to pathological consequences. Several studies have shown increased rates of colon cancer and prostate cancer associated with the intake of large doses of folic acid. However, the lack of folate in the diet, or the lack of folic acid supplementation, is directly correlated to neural tube defects occurring during fetal development.
The function of THF derivatives is to carry and transfer various forms of one carbon (1C) units during biosynthetic reactions. The one carbon units are either methyl, methylene, methenyl, formyl, or formimino groups.
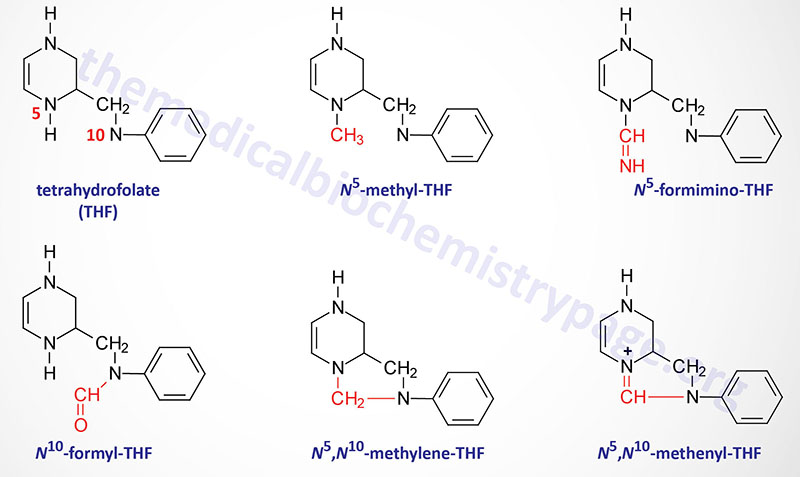
Reactions of Folate Derivative Synthesis
There are two distinct pools of THF molecules, the active pool and the reduced pool. The reduced pool consists solely ofN 5-methyl-THF (5-methyl-THF) which is also the major form of folate absorbed from the small intestines as well as the major form circulating in the blood for transport to the various tissues. All of the other THF derivatives constitute the active pool. The active pool forms of THF can be converted to 5-methyl-THF but 5-methyl-THF cannot be used, in any significant capacity, to contribute to the synthesis of the active pool folate derivatives. The majors pools of folate derivatives are found in the liver where they are distributed between the cytosol and the mitochondria. The majority of the mitochondrial pool of folate is THF and 10-formyl-THF with less than 10% being 5-methyl-THF and 5-formyl-THF. Within the cytosol nearly 50% of folate is in the 5-methyl-THF form with around 25% being THF and around 25%–30% being combined between the 10-formyl-THF and 5-formyl-THF forms.
N 5–N 10-Methylenetetrahydrofolate Synthesis
N 5,N 10-methylene-THF (5,10-methylene-THF) is synthesized from THF and serine by the serine/glycine hydroxymethyltransferases (SHMT1 and SHMT2). The SHMT1 encoded enzyme is cytosolic and the SHMT2 encoded enzyme is mitochondrial. The carbon from serine (specifically carbon 3) represents the major source of the one carbon (1C) units transferred during reactions utilizing THF. Indeed, the ability of serine to be synthesized from the glycolytic intermediate, 3-phosphoglycerate, directly ties glucose metabolism to the active THF pool. The SHMT catalyzed reactions generating 5,10-methylene-THF not only represent the major active THF synthesizing reaction but also represents a major reaction for the conversion of serine to glycine, a reaction that directly ties glycine synthesis to glycolysis. The 5,10-methylene-THF form of folate is required for the synthesis of thymidine nucleotides which directly ties DNA synthesis to glucose metabolism.
N 5-Methyltetrahydrofolate Synthesis
The conversion of 5,10-methylene-THF to 5-methyl-THF is catalyzed by methylene THF reductase (encoded by the MTHFR gene). Mutations in the MTHFR gene results in a form of homocysteinemia that is often referred to as hyperhomocysteinemia. The MTHFR gene is located on chromosome 1p36.22 and is composed of 12 exons that generate two alternatively spliced mRNAs, both of which encode distinct protein isoforms.
N 5-Formininotetrahydrofolate Synthesis
The synthesis ofN 5-formimino-THF (5-formimino-THF) occurs during the catabolism of histidine and is catalyzed by formimidoyltransferase cyclodeaminase (encoded by the FTCD gene). The FTCD gene is located on chromosome 21q22.3 which is composed of 16 exons that generate three alternatively spliced mRNAs, two of which encode the same 541 amino acid protein (isoform A) and the other encodes a protein of 572 amino acids (isoform C).
N 10-Formyltetrahydrofolate Synthesis
Synthesis ofN 10-formyl-THF (10-formyl-THF) can occur by two distinct pathways, both of which utilize the same multifunctional enzyme encoded by the MTHFD1 gene. There are three activities of the MTHFD1 encoded enzyme that include,N 5,N 10-methenyl-THF (5,10-methenyl-THF) cyclohydrolase, 5,10-methylene-THF dehydrogenase, and 10-formyl-THF synthetase. In one pathway 5,10-methenyl-THF is converted to 10-formyl-THF and in the other pathway 10-formyl-THF is formed from ATP, formate, and THF. The 10-formyl-THF form of folate is required for the synthesis of the purine nucleotides. The 5,10-methylene dehydrogenase activity of the MTHFD1 encoded enzyme converts 5,10-methylene-THF to 5,10-methenyl-THF. The MTHFD1 gene is located on chromosome 14q23.3 and is composed of 28 exons that generate two alternatively spliced mRNAs that encode distinct protein isoforms.
MTHFD1-Related Enzymes
Humans express additional genes that encode enzymes with activities similar the MTHFD1 encoded enzyme. The MTHFD2 gene encodes a bifunctional enzyme localized to the mitochondria. The MTHFD2 encoded enzyme is an NAD+/NADP+-dependent enzyme that possesses 5,10-methylene-THF dehydrogenase and 5,10-methenyl-THF cyclohydrolase activities. One critical function of the MTHFD2 encoded enzyme is in the mitochondrial catabolism of serine to maintain the NADH pool.
Two additional genes, MTHFD1L and MTHFD2L encode mitochondrial NADP+-dependent enzymes. The MTHFD1L encoded enzyme is a mono-functional enzyme with 10-formyl-THF synthetase activity and the MTHFD2L encoded enzyme is a bi-functional enzyme with 5,10-methylene-THF dehydrogenase and 5,10-methenyl-THF cyclohydrolase activities.
One-Carbon Transfer Reactions
The one carbon transfer reactions involving the various THF derivatives are required in the biosynthesis of serine, methionine, glycine, choline, the purine nucleotides, and dTMP. Indeed, the interconversion of serine and glycine, via the involvement of THF, represents a major pathway for the generation of 5,10-methylene-THF which of a member of the active folate pool. The active folate pool includes 10-formyl-THF, 5,10-methenyl-THF, and 5,10-methylene-THF. Methyl-THF is referred to as the reduced folate pool. All of the active folate pool molecules can be converted to 5-methyl-THF but the reverse process does not occur. For this reason, the inability to convert 5-methyl-THF to THF (as occurs via the B12– and folate-dependent enzyme, methionine synthase (also known as homocysteine methyltransferase) and then to any other THF derivative leads to trapping of folate in the "reduced" form.
The ability to acquire choline and amino acids from the diet and to salvage the purine nucleotides makes the role of 5,10-methylene-THF, in dTMP synthesis, one of the most significant metabolic functions for dietary folate. The role of vitamin B12 and 5-methyl-THF in the conversion of homocysteine to methionine also can have a significant impact on the ability of cells to regenerate the needed active pool of THF derivatives and as such represents the physiologically most significant reaction involving folate. Deficiencies in B12 or defects in methionine synthase will result in 5-methyl-THF trapping with the result being the development of megaloblastic anemia due to the block in nucleotide production and the inability of bone marrow erythroblasts to proceed through S-phase of the cell cycle.
Dietary Source of Folate
The recommended daily allowance for folic acid is 400 micrograms (μg) for both men and women except that pregnant women should increase their intake to at least 600 μg/day. The best sources for folic acid are cereals that have been fortified with 100% of the RDA of folic acid. Beef liver (85 gm) contains 45% of the RDA of folic acid. Excellent vegetarian sources for folic acid are baked beans, raw spinach, asparagus, green peas, broccoli, lentils, turnip greens, egg noodles, avocado, peanuts, lettuce, wheat germ, tomato juice and orange juice.
Clinical Significance of Folate Deficiency
Folate deficiency results in complications nearly identical to those described for vitamin B12 deficiency. The most pronounced effect of folate deficiency on cellular processes is upon DNA synthesis. This is due to an impairment in dTMP synthesis which leads to cell cycle arrest in S-phase of rapidly proliferating cells, in particular hematopoietic cells. The result is megaloblastic anemia as for vitamin B12 deficiency. The inability to synthesize DNA during erythrocyte maturation leads to abnormally large erythrocytes termed macrocytic (megaloblastic) anemia.
Since both folate and vitamin B12 deficiencies result in megaloblastic anemias, it is necessary to be able to clinically distinguish the cause as it relates to vitamin deficiency. Since B12 is required for both the methionine synthase reaction and the methylmalonyl-CoA mutase reaction a megaloblastic anemia resulting from B12 deficiency is also associated with an accompanying methylmalonic acidemia, whereas megaloblastic anemias caused by folate deficiency are not. An additional critical diagnostic is related to the onset of symptoms. Because human cells cannot store any significant amount of folate but the liver can store 3-6 years worth of B12, folate deficient megaloblastic anemias manifest very quickly, whereas B12 deficient anemias take considerably longer to manifest.
Folate deficiencies are rare due to the adequate presence of folate in food. Poor dietary habits as those of chronic alcoholics can lead to folate deficiency. The predominant causes of folate deficiency in non-alcoholics are impaired absorption or metabolism or an increased demand for the vitamin. The predominant condition requiring an increase in the daily intake of folate is pregnancy. This is due to an increased number of rapidly proliferating cells present in the blood. The need for folate will nearly double by the third trimester of pregnancy. Certain drugs such as anticonvulsants and oral contraceptives can impair the absorption of folate. Anticonvulsants also increase the rate of folate metabolism.
Inherited Defects in Folate Metabolizing Enzymes
Mutations in the methylene THF reductase gene (MTHFR) are associated with a disorder that was originally referred to as hyperhomocysteinemia. Characteristic features of hyperhomocysteinemia are a high plasma homocysteine level and an associated early onset of coronary artery disease (CAD). Additional symptoms of hyperhomocysteinemia include mild cognitive impairment and the potential for dementia. Indeed, there is evidence that links elevated serum homocysteine levels with Alzheimer disease. The most common mutation in the MTHFR gene resulting in this disorder is a C to T change at nucleotide position 677 (C677T).
Differential Diagnosis of Vitamin Deficiency-Induced Macrocytic Anemias
| Deficiency/Defect | Characteristics |
| B12 deficiency | B12 is required for the methionine synthase and methylmalonyl-CoA mutase catalyzed reactions; deficiency in this vitamin is associated with both homocysteinemia and megaloblastic (macrocytic) anemia; the megaloblastic anemia associated with B12 deficiency is the result of folate (asN 5-methyl-THF) trapping at the methionine synthase reaction; homocysteinemia results from the resultant inhibition of the methionine synthase reaction which leads to increased production of homocysteine which cannot be adequately accommodated in the catabolic direction at the cystathionine β-synthase (CBS) reaction; because methionine synthase is inhibited there will be areduction in measurable methionine in the blood and urine; the resultant homocysteinemia is associated with asignificant increase in the risk for cardiovascular disease such as deep vein thrombosis (DVT) and atherosclerosis; both the homocysteinemia and the megaloblastic anemia that result from B12 deficiency will be accompanied bydramatic increases in measurable methylmalonic acid in the serum which isNOT seen in folate deficiency; because the liver can store B12 for up to 6 years it takes a considerable amount of time for a dietary (or other cause) deficiency in B12 to manifest |
| Folate deficiency | folate is also required for the methionine synthase reaction and, therefore, as for B12 deficiency, there will be both homocysteinemia and megaloblastic anemia with deficiency in this vitamin; in the case of folate deficiency, the megaloblastic anemia results from a direct loss of purine nucleotide and thymidine nucleotide synthesis; folate deficiency is associated withreduction in measureable serum and urine methionine as for B12 deficiency; the resultant homocysteinemia results in asignificant increase in the risk for cardiovascular disease such as DVT and atherosclerosis as for B12 deficiency; since there is no associated inhibition of the methylmalonyl-CoA mutase reaction, there isno associated methylmalonic acidemia with the folate deficiency-associated homocysteinemia and megaloblastic anemia; because the human body cannot store folate, and the active folate pool is very small, the onset of symptoms associated with folate deficiency isvery rapid |
Ascorbic Acid: Vitamin C
Ascorbic acid is more commonly known as vitamin C. Ascorbic acid is derived from glucose via the uronic acid pathway, however, the enzyme L-gulono-γ-lactone oxidase responsible for the conversion of gulonolactone to ascorbic acid is absent in primates making ascorbic acid required in the diet. The gene encoding the enzyme (symbol: GULO) is present in the human genome but it lacks five of the twelve exons required to make a functional protein. Therefore, the gene is considered a pseudogene and the designation for the gene in humans is GULOP.
The active form of vitamin C is ascorbic acid itself. The main function of ascorbate is as a reducing agent in a number of different reactions. Ascorbate is the cofactor for Cu+–dependent monooxygenases and for several 2-oxoglutarate and Fe2+–dependent dioxygenases (2OG-oxygenases). Several critical enzymes that require ascorbate as a cofactor include the collagen processing enzymes, the lysyl hydroxylases and the prolyl hydroxylases as well as the catecholamine synthesis enzyme dopamine β-hydroxylase. Dietary ascorbate is also involved in non-heme iron absorption in the small intestine and in the overall regulation of iron homeostasis. In addition to its role in the regulation of the redox state of iron, ascorbate has the potential to reduce cytochromesa andc of the respiratory chain as well as molecular oxygen.
The most important reactions requiring ascorbate as a cofactor are the hydroxylations of lysine and proline residues in collagen. Vitamin C is, therefore, required for the maintenance of normal connective tissue as well as for wound healing since synthesis of connective tissue is the first event in wound tissue remodeling. Vitamin C also is necessary for bone remodeling due to the presence of collagen in the organic matrix of bones. Several other metabolic reactions require vitamin C as a cofactor. These include the catabolism of tyrosine, the synthesis of epinephrine from tyrosine (critical enzyme is dopamine β-hydroxylase), the synthesis of carnitine, peptide hormone amidation, and the synthesis of bile acids. It is also believed that vitamin C is involved in the process of steroidogenesis since the adrenal cortex contains high levels of vitamin C which are depleted upon adrenocorticotropic hormone (ACTH) stimulation of the gland.
Ascorbic acid also serves as a reducing agent and as an antioxidant. A critical anti-oxidant function of ascorbate is in the plasma membrane reduction of oxidized α-tocopherol (vitamin E). When functioning as an antioxidant, ascorbic acid itself becomes oxidized to semidehydroascorbate and then dehydroascorbate. Semidehydroascorbate is reconverted to ascorbate in the cytosol by cytochromeb5 reductase and thioredoxin reductase in reactions involving NADH and NADPH, respectively. Dehydroascorbate, the fully oxidized form of vitamin C, is reduced spontaneously by glutathione, as well as enzymatically in reactions using glutathione or NADPH.
Ascorbate is found at highest concentrations within nucleated cells with the intracellular concentration being found on the order of 30-100 times the concentration found in the plasma. The uptake on ascorbate from the lumen of the small intestine as well as its transport into cells is the function of two Na+-dependent vitamin C co-transporters identified as SVCT1 and SVCT2. Expression of SVCT1 is predominantly in intestinal epithelial cells and renal tubular cells where the transporter is found in the apical plasma membrane. Expression of SVCT2 is found in almost all nucleated cells. Expression of the SVCT2 transporter in the brain is critical for normal brain function and for ascorbate homeostasis within the brain. Both SVCT1 and SVCT2 are members of the SLC23 family of solute carriers and as such the gene encoding SVCT1 is SLC23A1 and that encoding SVCT2 is SLC23A2. Although the SVCT are the primary transporters for ascorbate, cells are also capable of ascorbate uptake via the activity of the facilitative glucose transporters, GLUT1, GLUT3, and GLUT4. Indeed, erythrocyte accumulation of ascorbate occurs primarily through the action of GLUT1.
Dietary Sources of Vitamin C
The amount of vitamin C that is recommended to consume each day (the RDA) depends upon the age and sex of the individual. Infants less than 1 year old should get 50 milligrams (mg) per day. children 1–3 years old need 15mg, 4–8 years old need 25mg, and 9–13 years old need 45mg. Adolescent girls should get 65mg per day and adolescent boys should get 75mg per day. Adult males need 90mg per day and adult women should get 75mg per day. Women who are breastfeeding should increase their intake to at least 120mg per day. Individuals who smoke should increase their daily intake by at least 35mg since smoking depletes vitamin C levels. The recommended daily intake of vitamin C to prevent conditions such as the cardiovascular disorders indicated above is reported to be between 500mg and 1000mg.
Excellent sources of vitamin C are fruits and vegetables such as oranges, watermelon, papaya, grapefruit, cantaloupe, strawberries, raspberries, blueberries, cranberries, pineapple, kiwi, mango, green peppers, broccoli, turnip greens, spinach, red and green peppers, canned and fresh tomatoes, potatoes, Brussels sprouts, cauliflower, and cabbage. Citrus juices or juices fortified with vitamin C are also excellent sources of the vitamin.
Vitamin C is sensitive to light, air, and heat, so the most vitamin C is available in fruits and vegetables that are eaten raw or lightly cooked. Natural or synthetic vitamin C can be found in a variety of forms. Tablets, capsules, and chewables are probably the most popular forms, but vitamin C also comes in powdered crystalline, effervescent, and liquid forms. An esterified form of vitamin C is also available, which may be easier on the stomach for those who are prone to heartburn. The best way to take vitamin C supplements is 2–3 times per day, with meals, depending on the dosage.
Ascorbic Acid and Iron Homeostasis
Iron homeostasis is critical for cell survival. Indeed, iron-depletion rapidly results in cellular death. The details of dietary uptake and cellular storage and utilization of iron are presented in the Iron and Copper Homeostasis page, thus, this discussion will be limited to the role of ascorbate in the overall processes. As a brief overview, the adult human body contains 3–5 grams of iron. Of this, greater than 80% is utilized by red blood cells and is found within the hemoglobin in these cells. The vast majority of the remainder, approximately 20% of the total, is transiently stored within the liver and macrophages, with the liver being the major storage location. When inside cells, the bulk of iron is stored bound within complexes of the protein ferritin. The remainder of the iron is in the heme of cytochromes, iron-sulfur center (ISC)-containing proteins, and in non-heme and non-ISC iron-containing proteins.
When contained in proteins, iron is involved in redox reactions that are one-electron transfers between its ferric (Fe3+) and ferrous (Fe2+) forms. These electron transfers involving iron are vital for cell survival, but must be limited to the context of reactions directed by specific enzymatic reactions. Iron that is not bound in a redox-inert form is capable of generating highly deleterious reactive oxygen species (ROS) via a reaction known as the Fenton reaction. The product of this process is primarily the hydroxy free radical, the most poisonous of all ROS. Thus, as might be expected, iron homeostasis is tightly controlled through processes that regulate its import, storage and cellular efflux. Ascorbate is an essential co-factor in the overall process of iron homeostasis. Ascorbate stimulates dietary iron absorption, contributes to plasma transferrin-bound iron uptake following binding of the transferrin-iron complexes to the plasma membrane transferrin receptor, stimulates the synthesis of the iron storage protein ferritin, inhibits lysosomal ferritin degradation, and inhibits cellular iron efflux.
The absorption of non-heme iron, from the lumen of the small intestine, requires that the dietary iron, which is predominantly in the oxidized state (Fe3+), be reduced to the ferrous (Fe2+) state. Ferrous iron is then taken up into intestinal enterocytes via the action of the divalent metal transporter 1 (DMT1) protein. There are at least three mechanisms known to exist that contribute to the process of dietary iron reduction, all of which involve ascorbic acid.
The major mechanism for intestinal iron reduction involves a ferrireductase that is a member of the cytochrome b561 class of transmembrane redox enzymes. This intestinal ferrireductase is known as duodenal cytochromeb 561 (DCYTB) which is encoded by the CYBRD1 (cytochromeb reductase 1) gene. During the process of DCYTB-mediated reduction of lumenal ferric iron, intracellular ascorbate is oxidized to an ascorbyl radical and then to dehydroascorbate.
Ascorbic acid, as an ascorbyl radical within the lumen of the gut is also capable of reducing ferric iron to ferrous iron which can then be absorbed via DMT1. A third mechanism for intestinal ferric iron reduction involves ascorbic acid being oxidized to dehydroascorbate in the lumen of the intestine concomitant with reduction of ferric to ferrous iron. The resulting ferrous iron is absorbed by the enterocyte via DMT1 while the dehydroascorbate is transported into the enterocyte most likely via the action of GLUT1.
When stored within cells, iron is complexed with a heteromeric protein called ferritin. Ferritin, that stores iron is composed of 24 subunits, that include heavy ferritin (H-ferritin) and light ferritin (L-ferritin). When stored inside ferritin, the ferrous iron that was transported into the cell is oxidized back to the ferric state. The ferroxidase activity of ferritin is associated with H-ferritin. When ferritin stored iron is released it is reduced back to the ferrous state. This intracellular reduction process involves ascorbic acid.
Ferrous iron can be transported into the circulation from intestinal enterocytes and from hepatocytes which represent the major iron storage cells in the body. This transport involves the transmembrane transporter identified as ferroportin. Associated with ferroportin in the membrane of intestinal enterocytes is the ferroxidase called hepheastin which oxidizes the ferrous iron to the ferric state. When iron is transported from hepatocytes to the blood via ferroportin action, the ferrous iron is oxidized by the major plasma copper-dependent ferroxidase, ceruloplasmin.
The oxidation of ferrous iron to ferric is required for plasma transport as the major plasma iron transport protein, transferrin, only binds the ferric state iron. The iron bound to transferrin is taken up by most cells after binding of the transferrin-iron complexes to the plasma membrane transferrin receptor followed by endocytosis of the ligand-receptor complex. The internalization and release of iron and its reduction to the ferrous state within the cell involves intracellular ascorbic acid.
Clinical Significance of Ascorbate Deficiency
Deficiency in vitamin C leads to the diseasescurvy due to the role of the vitamin in the post-translational modification of collagens. Scurvy is characterized by easily bruised skin, muscle fatigue, soft swollen gums, decreased wound healing and hemorrhaging, osteoporosis, and anemia. Due to its role in collagen processing, the bleeding dysfunction associated with vitamin C deficiency is characterized by the lack of effect on prothrombin time (PT) but with a prolonged bleeding time. The latter effect is the result of the reduced ability for platelets to adhere to exposed sub-endothelial extracellular matrix collagen which is required for their activation.
Vitamin C is readily absorbed and so the primary cause of vitamin C deficiency is poor diet and/or an increased requirement. The primary physiological state leading to an increased requirement for vitamin C is severe stress (or trauma). This is due to a rapid depletion in the adrenal stores of the vitamin. The reason for the decrease in adrenal vitamin C levels is unclear but may be due either to redistribution of the vitamin to areas that need it or an overall increased utilization.
Inefficient intake of vitamin C has also been associated with a number of conditions, such as high blood pressure, gallbladder disease, stroke, some cancers, and atherosclerosis (plaque in blood vessels that can lead to heart attack and stroke). Sufficient vitamin C in the diet may help reduce the risk of developing some of these conditions, however, the evidence that taking vitamin C supplements will help or prevent any of these conditions is still lacking.
Vitamin A
Vitamin A consists of three biologically active molecules, retinol, retinal (retinaldehyde) and retinoic acid.
Each of these vitamin A compounds is derived from the plant precursor molecule, β-carotene (a member of a family of molecules known as carotenoids). Beta-carotene, which consists of two molecules of retinal linked at their aldehyde ends, is also referred to as the provitamin form of vitamin A. Additional provitamin A carotenoids are α-carotene and β-cryptoxanthin. Whereas β-carotene is a source of two moles of retinaldehyde (and thus two moles of retinol), α-carotene and β-cryptoxanthin provide only a single mole each of retinol. Thus, β-carotene represents the most potent provitamin A molecule. Humans also acquire vitamin A from animal-derived foods in the form of long-chain acyl esters of retinol
Dietary retinol from animal-derived foods is released from the fatty acid esters through the actions of pancreatic lipases and the retinol is then taken up into intestinal enterocytes. Within the intestinal enterocytes retinol is re-esterified to saturated fatty acids (principally palmitic acid) via the action of the enzyme known as lecithin:retinol acyltransferase (LRAT). The retinyl esters are then packaged into chylomicrons which, via the intestinal lymphatic system, are eventually delivered to the blood. Retinyl esters can also be transported from enterocytes into the portal circulation via the action of the ABC family member transporter, ABCA1. The ABCA1 transporter is also involved in the efflux of cholesterol from cells to high density lipoproteins, HDL.
Ingested β-carotene can be cleaved in the lumen of the intestine by either of two β-carotene oxygenases (β-carotene-15,15′-monooxygenases) encoded by the BCO1 and BCO2 genes to yield two molecules of retinaldehyde (retinal). The BCO1 gene is located on chromosome 16q23.2 and is composed of 11 exons that encode a 547 amino acid protein. Expression of the BCO1 gene predominates in the small intestine. The BCO2 gene is located on chromosome 11q23.1 and is composed of 13 exons that generate five alternatively spliced mRNAs, each of which encode a distinct protein isoform.
Retinaldehyde is reduced to retinol by one of a family of enzymes called retinol dehydrogenases. The originally identified activity was called retinaldehyde reductase. Retinol dehydrogenases use NADPH as a cofactor in the reduction of retinaldehyde to retinol and use NAD+ as cofactor when oxidizing retinol to retinaldehyde. Humans express over 20 genes that encode proteins exhibiting alltrans– orcis-retinol dehydrogenase activity. However, not all are involved in the conversion of retinaldehyde to retinol or the reverse reaction. The retinol dehydrogenases that have been shown to be involved in retinaldehyde and retinol interconversions are members of the short-chain dehydrogenase/reductase (SDR) superfamily of enzymes.
Several members of the alcohol dehydrogenase family of enzymes can also interconvert retinaldehyde and retinol. Around 50% of ingested carotenoids are converted to retinol in this way within the lumen of the duodenum. The remainder of ingested carotenoids is transported into intestinal enterocytes through the action of the scavenger receptor identified as SR-BI. SR-BI is also the receptor, on the surface of hepatocytes, for HDL. Beta-carotene taken up into enterocytes in this way is then cleaved into retinaldehyde through the action of the β-carotene oxygenase encoded by the BCO1 gene. The released retinaldehyde is then reduced to retinol via one of the retinol dehydrogenases. As indicated earlier, retinol within the enterocytes is esterified to saturated fatty acids and packaged into chylomicrons.
The uptake of chylomicron remnants by the liver results in delivery of retinol to this organ for storage as a lipid ester within lipid droplets in hepatocytes. Transport of retinol from the liver to extrahepatic tissues occurs by binding of hydrolyzed retinol to retinol binding proteins, RBPs. The predominant plasma carrier of hepatic retinol is retinol binding protein 4 (RBP4). The retinol-RBP4 complex is then transported to the cell surface within the Golgi and secreted. While in the vasculature, the RBP4-retinol complexes interact with the protein transthyretin which prevents loss of retinol via renal glomerular filtration. The uptake of retinol, from the blood, occurs in response to the binding of RBP4-retinol complexes to the plasma membrane receptor identified as STRA6 (stimulated by retinoic acid 6). When bound to STRA6 the retinol is removed from RBP4 and transported across the membrane into the cell. Within extrahepatic tissues retinol is bound, primarily, to one of two RBPs, identified as RBP1 and RBP2. Within the eye, the major retinol binding protein is RBP3.
Dietary Sources of Vitamin A
Vitamin A is found in dark green and yellow vegetables and yellow fruits, such as broccoli, spinach, turnip greens, carrots, squash, sweet potatoes, pumpkin, cantaloupe, and apricots, and in animal sources such as liver, milk, butter, cheese, and whole eggs. When determining the amount of vitamin A to consume each day the RDA is expressed as retinol activity equivalents (RAE).
| Food source | Vitamin A content (mcg: μg RAE) |
| Organ meats, cooked, 85 gm | 1490–9126 |
| Sweet potato with peel (1 medium), baked | 1096 |
| Carrots, cooked from fresh, 65 gm | 671 |
| Spinach, cooked from frozen, 65 gm | 573 |
| Kale, cooked from frozen, 65 gm | 478 |
| Beet greens, cooked, 65 gm | 276 |
| Dandelion greens, cooked, 65 gm | 260 |
| Cantaloupe, raw, ¼ medium melon | 233 |
| Mustard greens, cooked, 65 gm | 221 |
| Red sweet pepper, cooked, 65 gm | 186 |
| Chinese cabbage, cooked, 65 gm | 180 |
Gene Control Exerted by Retinoic Acid
Retinol is converted to retinaldehyde via the action of two classes of oxidizing enzymes, cytoplasmic and microsomal. The cytoplasmic enzymes are alcohol dehydrogenases (ADH) of the medium-chain dehydrogenase/reductase (MDR) family. The microsomal enzymes are retinol dehydrogenases (RDH) of the short-chain dehydrogenase/reductase (SDR) family. Conversion of retinaldehyde to retinoic acid is catalyzed by a family of retinaldehyde dehydrogenases (RALDH) that are members of the aldehyde dehydrogenase 1 family of enzymes. The retinoic acid producing enzymes are RALDH1 (gene symbol: ALDH1A1), RALDH2 (gene symbol: ALDH1A2), and RALDH3 (gene symbol: ALDH1A3). All three RALDH are expressed in tissue specific and developmental stage specific patterns. The degradation of retinoic acid is catalyzed by enzymes of the cytochrome P450 (CYP) family, specifically CYP26A1, CYP26B1, and CYP26C1.
Within cells, retinoic acid (as well as the related compound 9-cis-retinoic acid) binds to specific receptor proteins that are members of the nuclear receptor family of steroid and thyroid hormone receptors. Prior to interaction with retinoic acid, the receptor protein is bound to retinoic acid response elements (RARE) in retinoic acid responsive genes. Following binding, the receptor-retinoic acid complex alters the transcriptional activity of the associated genes. Retinoic acid responsive genes are involved in growth and differentiation pathways during embryogenesis that control organogenesis, cranio-facial development, central nervous system patterning, and axial patterning. In this capacity retinoic acid is considered a hormone of the steroid and thyroid hormone superfamily. The retinoic acid receptors are abbreviated RAR and the related retinoid X receptors (RXR) bind 9-cis-retinoic acid.
Several genes, whose patterns of expression are altered by retinoic acid, are involved in the earliest processes of embryogenesis including the differentiation of the three germ layers, organogenesis and limb development. Within the adult, retinoic acid is involved in processes of reproduction, immunity, memory, and learning. To date, there are more than 530 genes that have been shown to be directly regulated (either positive or negative) via interaction with retinoic acid bound RAR or RXR. The canonical retinoic acid response element (RARE) in retinoic acid target genes is the direct repeat 5'–PuG(G/T)(T/A)CA–3′ separated by a spacer of five nucleotides. This response element is referred to as the DR5 element.
Vision and the Role of Vitamin A
Photoreception in the eye is the function of two specialized neuron types located in the retina; the rod and cone cells. The rod cells are utilized only for seeing at night During the day rod cells continuously release the excitatory neurotransmitter glutamate. The released glutamate binds to the synaptic membranes of specialized cells called bipolar cells. The glutamate receptor on bipolar cells is the mGluR6 metabotropic receptor. For more information on the glutamate receptors go to the Biochemistry of Nerve Transmission page. Activation of bipolar cells induces them to release the inhibitory neurotransmitter, GABA. Release of GABA, thus leads to inhibition of the optic nerve.
Both rod and cone cells contain a photoreceptor pigment in their membranes. The photosensitive compound of most mammalian eyes is a protein called opsin to which is covalently coupled an aldehyde of vitamin A. The opsin of rod cells is called scotopsin. The photoreceptor of rod cells is specifically called rhodopsin or visual purple. This compound is a complex between scotopsin and the 11-cis-retinal (also called 11-cis-retinene) form of vitamin A. Rhodopsin is a G-protein coupled receptor (GPCR) embedded in the membrane of the discs inside the outer segment of rod cells. Coupling of 11-cis-retinal occurs at three of the transmembrane domains of rhodopsin. Intracellularly, rhodopsin is coupled to a specific G-protein called transducin. The specific Gα-subunit of transduction is identified as Gαt.
Clinical Significance of Vitamin A Deficiency
Vitamin A is stored in the liver and deficiency of the vitamin occurs only after prolonged lack of dietary intake. As a fat soluble vitamin any lipid absorption disorder can be associated with deficiency in the vitamin. Patients with cystic fibrosis are particularly prone to deficiencies in the fat soluble vitamins due to defective pancreatic enzyme secretion and function. The earliest symptoms of vitamin A deficiency are night blindness. Additional early symptoms include follicular hyperkeratinosis, increased susceptibility to infection and cancer and anemia equivalent to iron deficient anemia. Prolonged lack of vitamin A leads to deterioration of the eye tissue through progressive keratinization of the cornea which results in abnormal dryness of the conjunctiva and cornea and the lack of ability of produce tears, a condition known asxerophthalmia.
The increased risk of cancer in vitamin A deficiency is thought to be the result of a depletion in β-carotene. Beta-carotene is a very effective antioxidant and is suspected to reduce the risk of cancers known to be initiated by the production of free radicals. Of particular interest is the potential benefit of increased β-carotene intake to reduce the risk of lung cancer in smokers. However, caution needs to be taken when increasing the intake of any of the lipid soluble vitamins. Excess accumulation of vitamin A in the liver can lead to toxicity which manifests as bone pain, hepatosplenomegaly, nausea and diarrhea.
Vitamin D
Biologically active vitamin D is a member of the steroid and thyroid hormone superfamily of lipophilic hormones. Vitamin D functions to regulate specific gene expression following interaction with its intracellular receptor, the vitamin D receptor (VDR). The VDR is a member of the steroid and thyroid hormone superfamily of nuclear receptors. The VDR gene is located on chromosome 12q13.11 and is composed of 12 exons that generate six alternatively spliced mRNAs. The VDRA mRNA represents the predominant transcript from the VDR gene and this mRNA encodes two protein isoforms that are the result of the use of alternative in-frame translation stop codons. The protein that results from the use of the upstream UGA stop codon is identified as VDRA or simply VDR. The protein that results from the use of the downstream UGA stop codon, due to readthrough translation, is identified as VDRAx. One of the other alternatively spliced mRNAs contains a 5′-non-coding exon but encodes the same VDRA protein.
The hormonally active form of vitamin D is 1,25-dihydroxy vitamin D3 (1,25-(OH)2D3, also called 1,25-dihydroxycholecalciferol or more commonly calcitriol). Calcitriol functions primarily to regulate calcium and phosphorous homeostasis. The transcriptional activity of the VDR is regulated not only by binding calcitriol but via the formation of heterodimers with the other nuclear receptors, specifically members of the RXR family. At sufficient concentration calcidiol, an intermediate in calcitriol synthesis, can also activate the VDR.
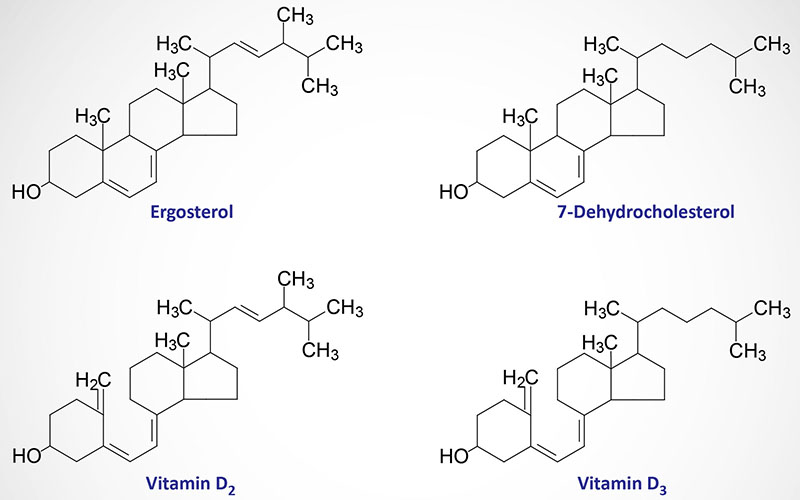
Active calcitriol is derived from ergosterol (produced in plants) and from 7-dehydrocholesterol which is an intermediate in the synthesis of cholesterol that accumulates in the skin. Upon exposure to ultraviolet B wavelength light (UVB: 280nm–315nm) from the sun (or from UVA wavelength light from tanning beds), 7-dehydrocholesterol is non-enzymatically (thermal isomerization) converted first to pre-vitamin D3 and then to vitamin D3. Ergocalciferol (vitamin D2) is formed by UVB irradiation of ergosterol. Both vitamin D2 and D3 enter the bloodstream and are taken up by the liver. Within the liver vitamins D2 and D3 undergo the first of two activating hydroxylation reactions. Vitamin D2 and D3, obtained from the diet (e.g. from fortified milk), are processed to D2-calcitriol and D3-calcitriol, respectively, by the same enzymatic pathways in the body as those that convert 7-dehydrocholesterol and ergosterol to their respective calcitriol forms.
Dietary vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) are absorbed from the intestine and transported to the liver bound to a specific vitamin D-binding protein which is a glycosylated α-globulin encoded by the GC gene (originally calledgroup-specificcomponent globulin). The GC gene is located on chromosome 4q13.3 and is composed of 15 exons that generate three alternatively spliced mRNAs that encoded two different protein isoforms.
In the liver cholecalciferols are hydroxylated at the 25 position by a specific vitamin D 25-hydroxylase generating 25-hydroxy-D3 [25-(OH)D3: more commonly called calcidiol]. The vitamin D 25-hydroxylase is a microsomal membrane localized enzyme encoded by the CYP2R1 (cytochrome P450 2R1) gene. The CYP2R1 gene is located on chromosome 11p15.2 and is composed of 9 exons that generate six alternatively spliced mRNAs encoding three distinct protein isoforms.
Conversion of cholecalciferols to calcidiol is dependent solely upon substrate availability as there is no regulatory mechanism to control the formation of calcidiol. Calcidiol is the major form of vitamin D found in the circulation and is transported in the blood bound to the vitamin D-binding protein encoded by the GC gene. Calcidiol is absorbed from the blood by numerous cells of the body with the kidney tubule cells being the most significant for the biological activity of vitamin D.
Conversion of calcidiol to its biologically active form, calcitriol, occurs through the activity of a specific 25-hydroxyvitamin D3 1-α-hydroxylase (commonly just called 1-α-hydroxylase) present in the inner mitochondrial membrane of cells of the proximal convoluted tubules of the kidneys, and in several extrarenal sites such as in keratinocytes, macrophages, and placenta.
All of the regulation in the formation of biologically active vitamin D occurs at the reaction catalyzing the synthesis of calcitriol from calcidiol. The 25-hydroxyvitamin D3 1-α-hydroxylase enzyme is encoded by the CYP27B1 (cytochrome P450 27B1) gene. The CYP27B1 gene is located on chromosome 12q14.1 and is composed of 9 exons that encode a 508 amino acid protein.
Calcidiol can also be hydroxylated at the 24 position by a specific vitamin D3-24-hydroxylase (encoded by the CYP24A1 gene) in the kidneys, intestine, placenta and cartilage. The 24,25-dihydroxycholecalciferol [24,25-(OH)2D3] product of the CYP24A1 gene is an inactive form of vitamin D and its generation represents a mechanism of regulating the circulating levels of calcitriol.
The primary functions of calcitriol are exerted in concert with parathyroid hormone (PTH) and involve the regulation whole body calcium and phosphorous homeostasis. PTH is released from the parathyroid gland in response to low serum calcium and induces the production of calcitriol by activating the expression of the renal CYP27B1 gene. Circulating calcium levels activate the calcium sensing receptor (CaSR) present in the parathyroid gland. Activation of the CaSR, when there are normal levels of serum calcium, results in increased proteolysis of PTH resulting in little secretion of the hormone from the parathyroid gland. However, in conditions of hypocalcemia the CaSR is not activated and parathyroid gland PTH is stable and secreted into the circulation. When there are reduced levels of PTH in the circulation the CYP24A1 gene is active leading to the stimulated synthesis of the inactive 24,25-(OH)2D3.
When PTH levels rise in the blood the hormone binds to its receptor on the surface of cells. There are two PTH receptors identified as PTH-1 and PTH-2. Within the kidney the PTH receptor is the PTH-1 isoform. When PTH-1 is activated by PTH binding there is enhanced synthesis and activity of the 25-hydroxyvitamin D3 1-α-hydroxylase enzyme. Activation of the 25-hydroxyvitamin D3 1-α-hydroxylase enzyme results in increased synthesis of calcitriol which is then released into the blood.
When taken up by cells, calcitriol functions as a steroid hormone inducing the expression of calcium-binding proteins called calbindins that facilitate calcium transport within cells. The original calcitriol induced calcium-binding protein was identified in chicken duodenal tissues and was later called calbindin-D28K. In humans, and other mammals, the intestinal calcitriol-induced gene is the S100G (also known as CALB3) gene which encodes calbindin-D9K. In humans, calbindin-D28K is encoded by the CALB1 gene which is expressed in the kidney tubule and is responsible for participating in renal Ca2+ reabsorption.
Like avian intestinal calbindin-D28K, mammalian intestinal calbindin-D9K binds the absorbed calcium at the apical membrane and transports it across the cell to the basolateral membrane where it can be transported into the blood. In addition to activating expression of the S100G (CALB3) gene in the intestine, calcitriol also enhances the expression of several intestinal calcium transporter genes including the TRPV6 gene (encoding the transient receptor potential vanilloid 6 transporter), the SLC8A1 gene (encoding the Na+-Ca2+ exchanger; NCX1), and the ATP2B1 gene (encoding the plasma membrane Ca2+ transporter 1b; PCMA1b). As indicated above, the steroid hormone action of vitamin D occurs via the action of calcitriol binding to a specific intracellular receptor that is a member of the nuclear receptor family of hormone receptors called the vitamin D receptor (VDR).
The overall effects of calcitriol on intestinal calcium absorption are of several different modes. This is due to the fact that intestinal calcium absorption occurs via both paracellular (between cells) and transcellular (through cells) mechanisms. Intestinal concentrations of calcium are much higher in the lumen than in the enterocyte which establishes a gradient favoring passive transcellular movement of calcium. Transcellular calcium movement from the apical membrane to the basolateral involves the calmodulin-actin-myosin complex I. When the intracellular concentration of calcium rises this transport process is less favorable since the calcium gradient is reduced. The transcellular transport of intestinal calcium under these conditions is enhanced by calcitriol activating the expression of the calcium transporter genes just described.
The paracellular transport of intestinal calcium is indirectly influenced by calcitriol activating PKC which phosphorylates tight junction proteins making them more permeable to calcium. The increased absorption of calcium ions requires concomitant absorption of a negatively charged counter ion to maintain electrical neutrality. The predominant counter ion is inorganic phosphate (Pi) which is also taken up from the intestine by both paracellular and transcellular mechanisms.
When plasma ionized calcium (Ca2+) levels fall, the levels of PTH and calcitriol rise and their major sites of concerted action are bone, where they stimulate bone reabsorption, and the kidneys where they inhibit calcium excretion by stimulating reabsorption at the distal convoluted tubule and the collecting ducts.
Intestinal phosphate ion uptake from the lumen is the function of the SLC34A2 gene encoded transporter identified as NPT2b (Na+-phosphate cotransporter 2b). Hormonally active vitamin D also plays a role in intestinal phosphate uptake through calcitriol regulation of the expression of the SLC34A2 gene. Within the kidneys calcitriol increases phosphate reabsorption in the proximal tubule which counters the effects of PTH. Due to the fact that changes in calcitriol levels alter plasma calcium and PTH levels it is not clear what the exact mode of action is for calcitriol relative to renal phosphate retention.
Dietary Sources of Vitamin D
Due to the critical role of vitamin D in the development of growing bone as well as the maintenance of healthy bone it has been added to milk since the 1930's. In the US all milk is fortified with 100 IU/8 ounces (236 ml). Fatty fishes such as salmon, sardines and tuna are also an excellent source of vitamin D. Cod liver oil is also very high in vitamin D. For vegetarians and vegans ultraviolet (uv) light irradiated mushrooms and yeast are the only sources of vitamin D that are not animal in origin. The irradiation of yeasts and mushrooms produces vitamin D2. Vitamin D content is listed as the amount in International Units (IU).
| Food source | Vitamin D content (IU) |
| Cod liver oil, 15ml | 1360 |
| Salmon, cooked, 100 gm | 447 |
| Sardines, canned in oil, 50 gm | 250 |
| Tuna, canned in water, 85 gm | 154 |
| Egg, 1 large | 41 |
| Swiss cheese | 6 |
Although calcitriol is a major hormone involved in, primarily, calcium and phosphorous homeostasis, the VDR is found widely distributed in numerous different cells throughout the body. Many of the tissues that express the VDR gene have no role in calcium and phosphate homeostasis. Therefore, the presence of the VDR in these tissues indicates that calcitriol has functions in more cellular processes or that there are ligands other than calcitriol that can bind to and activate the VDR. Indeed, there are a large number of human genes that possess vitamin D-response elements (VDRE) that can bind the VDR and, therefore, be responsive to calcitriol. Calcitriol is, thus, capable of being involved in processes such as the regulation of cell proliferation, cell differentiation, and apoptosis. Several lines of evidence demonstrate that calcitriol exerts immunomodulatory and anti-proliferative effects and these effects have been hypothesized to demonstrate a role for calcitriol as a potential preventative or therapeutic in chronic auto-immune conditions such as type 1 diabetes.
Clinical Significance of Vitamin D
Vitamin D Deficiency
As a result of the addition of vitamin D to milk, deficiencies in this vitamin are rare in most developed countries. As a fat soluble vitamin, any lipid absorption disorder can be associated with deficiency in the vitamin. Patients with cystic fibrosis are particularly prone to deficiencies in the fat soluble vitamins due to defective pancreatic enzyme secretion and function. The main symptom of vitamin D deficiency in children isrickets and in adults isosteomalacia. Rickets is characterized by improper mineralization during the development of the bones resulting in soft bones. Osteomalacia is characterized by demineralization of previously formed bone leading to increased softness and susceptibility to fracture.
The resultant decrease in Ca2+ uptake due to vitamin D deficiency leads to hypocalcemia leading to excess parathyroid hormone release (referred to as secondary hyperparathyroidism) as a compensatory mechanism. The excess release of parathyroid hormone (PTH) can lead to over resorption of renal calcium leading to hypercalcemia. This hypercalcemia can cause calcification in soft tissues and peritrabecular fibrosis in bones. The peritrabecular fibrosis, associated with hyperparathyroidism, is quite distinct and is clinically called osteitis fibrosa cystica (OFC). OFC is also called von Recklinghausen disease of bone. This latter name is distinct from von Recklinghausen disease which is neurofibromatosis type 1.
Vitamin D Toxicity
Although vitamin D is critical to the regulation of calcium and phosphorous homeostasis it is possible to consume too much of the vitamin which results in vitamin D toxicity. Although supraphysiological dosing of vitamin D [considered to be any dose above 50,000 IU (2,500μg) per day for a duration of at least 1 month; normal adult RDA for vitmain D is 600 IU/day], and the associated toxicities are rare, it is a valid consideration in the context of the diagnosis of the causes of hypercalcemia. Both vitamin D2 and D3 (the cholecalciferols) are lipophilic and are rapidly taken up into skeletal muscle and adipose tissue where they can remain for several months. Given that the cholecalciferols are converted to calcidiol in the liver based solely on substrate availability, calcidiol will also accumulate under conditions of excess vitamin D intake. Calcidiol is also lipophilic and can be stored within adipose tissue for several months. Due to the storage of cholecalciferols and calcidiol in adipose tissue, vitamin D intoxication can take several weeks to resolve.
In the event of excessive vitamin D intake there is a very characteristic toxicity profile. The excess vitamin results in an associated hypercalcemia. The hypercalcemia caused by vitamin D toxicity is primarily due to the significant increases in circulating levels of calcidiol which, when it enters cells, can bind to and activate the VDR. In addition, at high concentrations calcidiol can displace calcitriol from vitamin D binding protein resulting in increased free calcitriol that can then activate the VDR. Both of these pathophysiological phenomena result in increased intestinal calcium absorption and increased bone calcium resorption. The resultant hypercalcemia can lead to pathologies that are similar to those caused by the hypercalcemia that results from the secondary hyperparathyroidism in the context of vitamin D deficiency.
One of the earliest tissues affected by the onset of the soft tissue calcification with vitamin D toxicity is the kidney. The symptoms that result from the damage to the renal tissue include polydipsia and polyuria. Additional symptoms of vitamin D toxicity include fatigue, nausea, vomiting, and diminished mental capacity. Chronic vitamin D toxicity can lead calcification of numerous soft tissues and may ultimately result in permanent renal failure.
Vitamin E
Vitamin E is a mixture of several related compounds known as tocopherols and tocotrienols. The tocopherols are the major sources of vitamin E in the U.S. diet. The tocopherols differ by the number and position of methyl (–CH3–) groups present on the ring system of the chemical structure. The different tocopherols are designated α-, β-, γ-, and δ-tocopherol. Most vitamin E in U.S. diets is in the form of γ-tocopherol from soybean, canola, corn, and other vegetable oils. All four tocopherols are able to act as free radical scavengers thus they all have potent antioxidant properties. Vitamin E is absorbed from the intestines packaged in chylomicrons. It is delivered to the tissues via chylomicron transport and then to the liver through chylomicron remnant uptake. The liver can export vitamin E in very low density lipoproteins (VLDLs). Within the liver α-tocopherol transfer protein preferentially transfers α-tocopherol to VLDLs, thus α-tocopherol is the most abundant tocopherol in non-hepatic (liver) tissues. Due to its lipophilic nature, vitamin E accumulates in cellular membranes, fat deposits and other circulating lipoproteins. The major site of vitamin E storage is in adipose tissue.
The major function of vitamin E is to act as a natural antioxidant by scavenging free radicals and molecular oxygen. In particular vitamin E is important for preventing peroxidation of polyunsaturated membrane fatty acids. The vitamins E and C are interrelated in their antioxidant capabilities. Active α-tocopherol can be regenerated by interaction with vitamin C following scavenge of a peroxy free radical. Alternatively, α-tocopherol can scavenge two peroxy free radicals and then be conjugated to glucuronate for excretion in the bile.
Although α-tocopherol is the most abundant tocopherol in tissues outside of the liver, it is not the most potent antioxidant form of the vitamin. Because of the unmethylated carbons in the ring structure of γ- and δ-tocopherol, these two forms of vitamin E are much more active at trapping free radicals, in particular reactive nitrogen species. In addition, research has recently shown that the anticancer effects of vitamin E are due to the γ- and δ-tocopherol forms and is not associated with α-tocopherol.
Dietary Sources of Vitamin E
Recommended daily intake amounts for vitamin E (as α-tocopherol) are listed in milligram (mg) amounts and also in International Units (IU). To convert from mg to IU: 1 mg of α-tocopherol is equivalent to 1.49 IU of the natural form or 2.22 IU of the synthetic form. Nuts, seeds, and vegetable oils are among the best sources of α-tocopherol, and significant amounts are available in green leafy vegetables and fortified cereals. Most vitamin E in US diets is in the form of γ-tocopherol from soybean, canola, corn, and other vegetable oils.
| Food source | Vitamin E content (mg) |
| Wheat germ oil, 15 ml | 20.3 |
| Almonds, dry roasted, 28 gm | 7.4 |
| Sunflower oil, 15 ml | 5.6 |
| Safflower oil, 15 ml | 4.6 |
| Peanut butter, 30 ml | 2.9 |
| Peanuts, dry roasted, 28 gm | 2.2 |
| Corn oil, 15 ml | 1.9 |
| Broccoli, chopped, boiled, 65 gm | 1.2 |
| Soybean oil, 15 ml | 1.1 |
| Kiwifruit, 1 medium | 1.1 |
| Spinach, raw, 130 gm | 0.6 |
Clinical Significance of Vitamin E Deficiency
Due to adequate vitamin E intake in the average American diet, no major deficiency syndromes are common. However, conditions that result from vitamin E deficiency are related to disturbances in nerve cell membrane lipid homeostasis. These conditions include cerebellar ataxias, myopathies, retinopathy, loss of deep tendon reflexes, and dysarthria (speech problem associated with difficulty articulating due to defects in the motor component of speech). Another major symptom of vitamin E deficiency in humans is an increase in red blood cell fragility due to the increased level of erythrocyte membrane lipid peroxidation in the absence of tocopherols. Since vitamin E is absorbed from the intestines in chylomicrons, any fat absorption disorder can lead to deficiencies in vitamin E intake.
Patients with cystic fibrosis are particularly prone to deficiencies in the fat soluble vitamins due to the defective pancreatic enzyme secretion and function. Increased intake of vitamin E is recommended in premature infants fed formulas that are low in the vitamin as well as in persons consuming a diet high in polyunsaturated fatty acids. Polyunsaturated fatty acids tend to form free radicals upon exposure to oxygen and this may lead to an increased risk of certain cancers.
Vitamin K
The K vitamins exist naturally as K1 (phylloquinone) in green vegetables and K2 (menaquinone) produced by intestinal bacteria and K3 is synthetic menadione. When administered, vitamin K3 is alkylated to one of the vitamin K2 forms of menaquinone.
The major function of the K vitamins is in the maintenance of normal levels of the blood clotting proteins, factors II, VII, IX, X and protein C and protein S, which are synthesized in the liver as inactive precursor proteins. Conversion from inactive to active clotting factor requires a posttranslational modification of specific glutamate (E) residues. This modification is a carboxylation and the enzyme responsible, γ-glutamyl carboxylase (GGCX), requires vitamin K as a cofactor. The GGCX gene is located on chromosome 21p11.2 and is composed of 15 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
The resultant modified E residues are γ-carboxyglutamate ( gla ). This process is most clearly understood for factor II, also called preprothrombin. Prothrombin is modified preprothrombin. Thegla residues are effective calcium ion chelators. Upon chelation of calcium, prothrombin interacts with phospholipids in membranes and is proteolyzed to thrombin through the action of activated factor X (Xa).
During the carboxylation reaction reduced hydroquinone form of vitamin K is converted to a 2,3-epoxide form. The regeneration of the hydroquinone form requires the action of the enzyme vitamin K epoxide reductase (VKORC1) which involves a two-step reaction. These latter reactions are the site of action of the coumarin based anticoagulants such as warfarin (trade name: Coumadin®). The VKORC1 gene is located on chromosome 16p11.2 and is composed of 4 exons that generate three alternatively spliced mRNAs, each of which encode a distinct protein isoform.
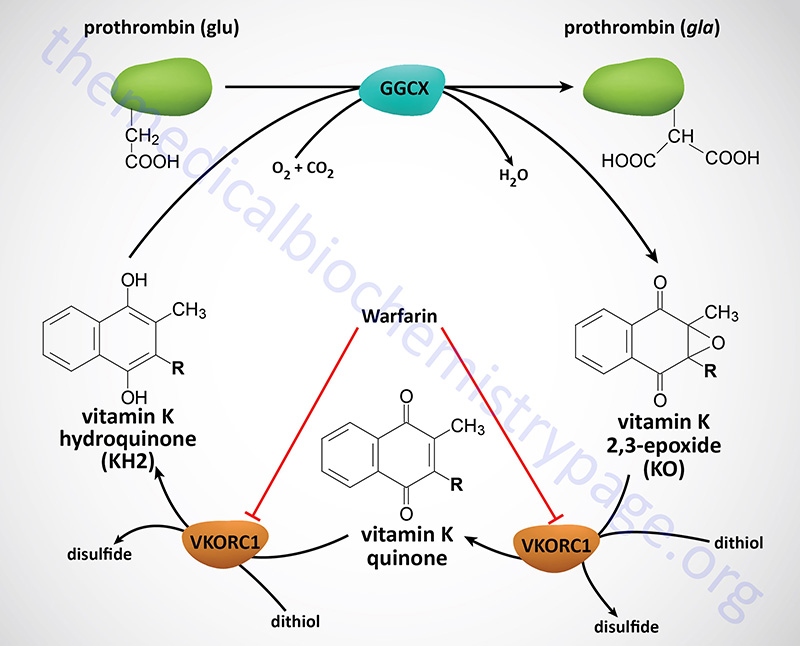
Dietary Sources of Vitamin K
In the average U.S. diet, meats and eggs are the most common food sources of the menaquinone form of vitamin K. Excellent sources of vitamin K include spinach, Brussels sprouts, Swiss chard, green beans, asparagus, broccoli, kale, mustard greens, green peas and carrots. Fermentation of foods can increase their vitamin K content. Fermented soy foods play a unique role in supplying vitamin K in certain traditional cuisines (like that of Japan). Sometimes you will see the word "natto" being used to refer to these fermented soy foods sinceBacillus natto are bacteria that can convert vitamin K1 into K2 and are often used in the production of fermented soy products. Some cheeses are also fermented in a way that optimizes their vitamin K content.
Clinical Significance of Vitamin K Deficiency
Naturally occurring vitamin K is absorbed from the intestines only in the presence of bile salts and other lipids through interaction with chylomicrons. Therefore, fat absorption disorders can result in vitamin K deficiency. Patients with cystic fibrosis are particularly prone to deficiencies in the fat soluble vitamins due to defective pancreatic enzyme secretion and function. The synthetic vitamin K3 is water soluble and absorbed irrespective of the presence of intestinal lipids and bile.
Since the vitamin K2 form is synthesized by intestinal bacteria, deficiency of the vitamin in adults is rare. However, long term antibiotic treatment can lead to deficiency in adults. The intestine of newborn infants is sterile, therefore, vitamin K deficiency in infants is possible if lacking from the early diet. The primary symptom of a deficiency in infants is a hemorrhagic syndrome.
Since vitamin K is critical for the generation ofgla residues in clotting factors, as described above, deficiency in this vitamin results in dysfunction in the processes of blood coagulation. Since the defect will be at the level of factors II, VII, IX, X and protein C and protein S, the effects of vitamin K deficiency can be seen in the context of the prothrombin time (PT) assay. Whereas, vitamin C deficiency is associated with bleeding dysfunction at the level of platelet adhesion to collagen and is, therefore, measurable with an associated increase in bleeding time, vitamin K deficiency has no effect on bleeding time.
Vitamin K Deficiency in the Neonate
Neonates are prone to vitamin K deficiency due to the limited levels in their bodies at birth and also to limited intake. As a result newborn infants are prone to a condition referred to as vitamin K deficiency-related bleeding (VKDB). Neonatal VDKB is defined as a bleeding disorder in which the coagulation deficit is rapidly corrected by vitamin K supplementation. The diagnosis of VDKB in an infant is indicated when the prothrombin time (PT) is found to be at least 4 times longer than the control value even though the infants platelet count and fibrinogen levels are normal. In order to confirm a diagnosis of VKDB one must measure for the level of the specific vitamin K-dependent factors (prothrombin, VII, IX, X). The level of these factors can be rapidly normalized via parenteral administration of 1 mg vitamin K.
There are various forms of VKDB that are defined by the age of onset (early, classic, and late) and as either idiopathic and secondary. Idiopathic VKDB is defined by the fact that no cause other than breast feeding can be determined. Secondary VKDB is associated with an underlying cause that can include an hereditary hepatobiliary or malabsorption disease (e.g. cystic fibrosis) or can be due to the effect of drugs that have been given to the mother or infant. In addition, autosomal recessive vitamin K-dependent coagulation factor deficiencies (VKCFD) may be the underlying cause of the VKDB. Inherited mutations in the γ-glutamyl carboxylase (GGCX) gene are the cause of VKCFD type I and mutations in the vitamin K epoxide reductase (VKORC1) gene are the cause of VKCFD type II.
Early onset VKDB presents within 24 hours of birth and is almost exclusively seen in infants of mothers taking drugs which inhibit vitamin K. These drugs include anticonvulsants (e.g. phenytoin, carbamazepine, or barbiturates), anti-tuberculosis drugs (e.g. rifampicin), certain antibiotics (e.g. cephalosporins), and vitamin K antagonists (e.g. warfarin). The incidence of early VKDB varies from 5%–10% in neonates of mothers taking these drugs. The VKDB in these cases is usually severe. Classical VKDB occurs between 24 hours and 7 days of life and is associated with delayed or insufficient feeding. The VKDB in these cases is usually mild. Late VKDB is associated with infants who are exclusively fed by the breast and occurs between 2 and 12 weeks after birth. The VKDB in these cases is severe with a mortality rate of 20%. In fully breast-fed infants, who do not receive supplemental vitamin K at birth, the incidence of late VKDB is between 1/15,000 and 1/20,000.
The primary underlying cause of VKDB in infants is related to the fact that their hemostatic system does not fully mature until 3 to 6 months of age. In addition, the amount and activity of the vitamin K-dependent procoagulant factors (prothrombin, VII, IX, and X) can be dramatically reduced due to insufficient storage and poor transfer of vitamin K across the placental barrier. Although the vast majority of neonates do not experience coagulation deficit, it is now generally considered routine to administer a prophylactic dose vitamin K at birth in order to prevent classical and late VKDB. Intramuscular administration of vitamin K has been universally adopted as the means of delivery. Nonetheless, it is usual and routine for continued oral administration of vitamin K.
Alpha-Lipoic Acid, LA
Alpha-lipoic acid, LA, (chemical name: 1,2-dithiolane-3-pentanoic acid; also known as thioctic acid), is a naturally occurring dithiol compound synthesized enzymatically in the mitochondrion from the medium-chain fatty acid octanoic acid. LA synthesis in humans is carried out by lipoyl (octanoyl) transferase 2 (encoded by the LIPT2 gene) and lipoic acid synthetase (encoded by the LIAS). The LIPT2 gene is located on chromosome 11q13.4 and is composed of 2 exons that as a result of the use of alternative splice sites generates three mRNAs, each of which encode distinct protein isoforms. The LIAS gene is located on chromosome 4p14 and is composed of 11 exons that generate six alternatively spliced mRNAs, each of which encodes a distinct protein isoform.
Because LA can be synthesized in the body it is not technically considered a vitamin but because of its vital role in overall cellular metabolism it is considered as an important, but not necessary, dietary supplement. Lipoic acid has one chiral center and therefore exists in bothR– andS-enantiomeric forms (see the Figure below). However, onlyR-LA is conjugated to conserved lysine residues in an amide linkage (forming a lipoamide), thus making this isoform essential as a cofactor in biological systems. Enzymes containing lipoamide are typically mitochondrial multi-enzyme complexes that catalyze the oxidative decarboxylation of α-keto acids such as the pyruvate dehydrogenase complex (PDHc), 2-oxoglutarate dehydrogenase (also known as α-ketoglutarate dehydrogenase), and branched-chain keto acid dehydrogenase (BCKD), and the glycine cleavage complex (GCC).
Given that LA is a necessary cofactor for mitochondrial α-ketoacid dehydrogenases, it thus serves a critical role in mitochondrial energy metabolism. In addition tode novo synthesis, LA is also absorbed intact from dietary sources, and it transiently accumulates in many tissues. LA has been described as a potent biological antioxidant, a detoxification agent, and a diabetes medicine. LA has been used to improve age-associated cardiovascular, cognitive, and neuromuscular deficits, and has been implicated as a modulator of various inflammatory signaling pathways. Accumulating evidence indicates that LA, supplied as a supplement in the diet, may not be used as a metabolic cofactor but instead, elicits a unique set of biochemical activities with potential therapeutic value against a host of pathophysiological insults.
Lipoic acid, either as a dietary supplement or a therapeutic agent, modulates distinct redox circuits because of its ability to equilibrate between different subcellular compartments as well as extracellularly. Because of its role in regulating redox states, LA is a critical component of the antioxidant network. It is important in the regeneration of other antioxidants, such as vitamins E and C, it increases the intracellular levels of glutathione (GSH), and provides redox regulation of numerous proteins and transcription factors. The extracellular thiol/disulfide red-ox environment (determined by the interconversion between cysteine and cystine) is able to modulate cell proliferation, apoptosis, cell adhesion molecules, and proinflammatory signaling.
Lipoic acid may play an important role in the modulation of the extracellular red-ox state via the involvement of dihydrolipoic acid (DHLA) in the reduction of cystine to cysteine. This reduction facilitates a rapid uptake of the cysteine into the cell making it available to stimulate GSH synthesis. Cellular uptake of LA has been shown to occur via several systems including the medium chain fatty acid transporter, a Na+-dependent vitamin transport system, and a H+-linked monocarboxylate transporter for intestinal uptake. The cellular reduction of LA to DHLA is accomplished by NAD(P)H-driven enzymes, thioredoxin reductase, lipoamide dehydrogenase, and glutathione reductase.

Lipoic acid is an essential cofactor for the E2 component of α-ketoacid dehydrogenase complexes that are exclusively located in mitochondria. These complexes include the pyruvate dehydrogenase complex (PDHc), 2-oxoglutarate (α-ketoglutarate) dehydrogenase (OGDH), and branched chain α-ketoacid dehydrogenase (BCKD) complexes. The PDHc catalyzes the oxidative carboxylation of pyruvate which serves as the entry point for carbohydrates into the TCA cycle as acetyl-CoA. OGDH catalyzes the conversion of 2-oxoglutarate (α-ketoglutarate) to succinyl-CoA in the TCA cycle. Evidence demonstrates that the activities of both PDH and OGDH are substantially decreased during aging and in neurodegenerative disorders.
Extensive evidence suggests that LA may have therapeutic usefulness in lowering blood glucose levels in diabetic conditions and that the intracellular redox status plays a role in the modulation of insulin resistance. Lipoic acid has been shown to stimulate glucose uptake by affecting components of the insulin signaling pathway. The signaling networks of insulin receptor activation include the insulin receptor substrates (IRS1, IRS2, and IRS4), PI3K, and PKB/AKT. There are three members of the PKB/AKT family of serine/threonine kinases identified as AKT1 (PKB, also PKBα), AKT2 (PKBβ), and AKT3 (PKBγ). Insulin-mediated activation of PI3K and PKB/AKT is necessary for the translocation of GLUT4 from an intracellular pool to the plasma membrane to allow for uptake of plasma glucose. Lipoic acid has been shown to augment tyrosine phosphorylation and the activity of components of insulin signaling including the insulin receptor, IRS1, PI3K, PKB/AKT, and p38MAPK. In addition, LA stimulated glucose uptake upon translocation and regulation of the intrinsic activity of GLUT4, an effect that is likely mediated by p38 MAPK.R-LA, as well as oxidized isoforms have been shown to stimulate glucose transport in differentiated 3T3-L1 adipocytes by a mechanism entailing changes in the intracellular redox status. These effects of LA are in agreement with an alteration of the thiol reactivity of redox components of the insulin signaling pathway caused by a thiol/disulfide exchange mechanism.
The stress-activated MAPK and JNK pathways play a central role in the progression of insulin resistance and diabetic neuropathies. The activation of these kinases results in the phosphorylation of IRS1 on serine 307 resulting in inhibition of the insulin-stimulated tyrosine phosphorylation of IRS1 leading to inhibition of insulin signaling. Lipoic acid has been shown to inhibit the JNK pathway and IRS1 serine phosphorylation, resulting in improved insulin sensitivity. The precise mechanism by which LA inhibits the JNK pathway remains unclear. Nevertheless, these effects place LA at the cross-road of insulin- and JNK signaling favoring glucose uptake and metabolism, thus ameliorating insulin resistance and improving the status of diabetes.
Source: https://themedicalbiochemistrypage.org/vitamins-water-and-fat-soluble/





0 Komentar